The authors describe the ways in which manufacturers can mitigate the risks related to the integrity of recombinant transgenes expressed in CHO cells.
By Luhong He, Christopher Frye
Abstract
 The Chinese hamster ovary (CHO) cell lines used for the production of recombinant therapeutic proteins are immortalized cells with a relatively high degree of genetic plasticity. Given this inherent genetic flux, recombinant genes (or transgenes, also referred to as the expression construct) expressed in CHO cells—used for therapeutic bioproduct development—can be subject to genetic alteration that may potentially impact the integrity and/or stability of those transgenes and, in turn, impact drug substance production. Provided is a comprehensive, risk-based transgene characterization strategy; its implementation is based on chemistry, manufacturing, and control (CMC) development phases to ensure that the integrity and stability of the transgene is maintained for clinical and commercial CHO production cell lines. Early-phase assessment includes characterization of the expression plasmid prior to cell-line generation (transfection); evaluation of transcript integrity of those transgenes expressed transiently and stably in CHO cells after transfection but prior to single-cell cloning of the candidate production cell lines using single-cell sorting (or alternative methods); and profiling of transgene copy number in cell-line populations across cell generations spanning the manufacturing window. Mid-phase assessment includes further characterization of the integrity and stability of the integrated transgenes using the defined commercial cell-culture processes. Finally, the presented strategy includes the late-phase characterization of the expression construct using cells at the limit for in vitro cell age harvested from the commercial cell-culture process to support the marketing authorization applications. Together, the presented strategy is integrated with other existing drug substance analytical control and product characterization strategies to ensure the integrity and consistency of the drug substance used for clinical and commercial applications.
The Chinese hamster ovary (CHO) cell lines used for the production of recombinant therapeutic proteins are immortalized cells with a relatively high degree of genetic plasticity. Given this inherent genetic flux, recombinant genes (or transgenes, also referred to as the expression construct) expressed in CHO cells—used for therapeutic bioproduct development—can be subject to genetic alteration that may potentially impact the integrity and/or stability of those transgenes and, in turn, impact drug substance production. Provided is a comprehensive, risk-based transgene characterization strategy; its implementation is based on chemistry, manufacturing, and control (CMC) development phases to ensure that the integrity and stability of the transgene is maintained for clinical and commercial CHO production cell lines. Early-phase assessment includes characterization of the expression plasmid prior to cell-line generation (transfection); evaluation of transcript integrity of those transgenes expressed transiently and stably in CHO cells after transfection but prior to single-cell cloning of the candidate production cell lines using single-cell sorting (or alternative methods); and profiling of transgene copy number in cell-line populations across cell generations spanning the manufacturing window. Mid-phase assessment includes further characterization of the integrity and stability of the integrated transgenes using the defined commercial cell-culture processes. Finally, the presented strategy includes the late-phase characterization of the expression construct using cells at the limit for in vitro cell age harvested from the commercial cell-culture process to support the marketing authorization applications. Together, the presented strategy is integrated with other existing drug substance analytical control and product characterization strategies to ensure the integrity and consistency of the drug substance used for clinical and commercial applications.
Over the past several decades, numerous recombinant proteins have been approved as therapeutic drugs by regulatory authorities, and many more are currently undergoing clinical development (1). Chinese hamster ovary (CHO)-derived cell lines have become the predominant host for the manufacturing of glycosylated therapeutic proteins. During this period of explosion in the application of CHO expression systems, considerable efforts have been made to improve recombinant protein production to meet the demands of high quantity and consistent quality of biopharmaceutical products. Those efforts can be categorized into two fundamental areas: improving therapeutic protein developability by protein engineering based on structure-activity relationship (SAR) studies, and improving therapeutic protein production systems with a focus on host-cell engineering,cell-culture medium, and process development.
While the aforementioned efforts successfully introduced clinical candidates with more desirable therapeutic traits (e.g., enhanced activity, chemical and physical stability) and boosted the productivity of CHO cell lines from < 100 mg/L to > 10g/L, unintended consequences have resulted from the extensively-engineered transgenes expressed in CHO hosts, which inherently possess a relatively high degree of genetic instability. The authors’ internal development experience, which is consistent with published literature, indicates that transgenes integrated into the CHO genome can, in some cases, result in unintended protein species caused by a number of potential mechanisms including transgene RNA (transcript) aberrant splicing (2), genetic mutation (3, 4), and amino acid misincorporation (5–7) during cell-culture processes. If not removed by the purification process, these unintended byproducts are typically considered product-related impurities (PRI), as defined by International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Q6B (8), and could have potential implications on the safety and efficacy of the intended products. In addition, the integrated transgenes may be partially or completely lost or silenced (through epigenetic mechanisms) during the CHO cell-culture process (9–13), which may potentially lead to a lack of robustness of the drug substance manufacturing process.
ICH Q5B (14) recommends that, “segments of the expression construct should be analyzed using nucleic acid techniques in conjunction with other tests performed on the purified recombinant protein for assuring the quality and consistency of the final product.” This manuscript presents a comprehensive genetic characterization strategy developed and implemented based on bioproduct chemistry, manufacturing, and control (CMC) development phases to ensure that the integrity and stability of the transgenes is maintained during the cell-culture process and in the cells at the limit for in vitro cell age. It is crucial to note that the genetic characterization strategy presented here is not an isolated practice but rather an important part of a holistic, integrated strategy assembled during bioproduct development, including an appropriate analytical control strategy coupled with extensive product characterization.
In addition to meeting the regulatory requirements that ensure the safety and efficacy of biopharmaceuticals for clinical and commercial applications, the strategy presented here also provides an approach that addresses business considerations. CMC development is a complex, expensive, and resource-intensive process. There are different strategies to addressing business needs. At one extreme would be initiation of early clinical development using drug substance from a manufacturing process that would have to change significantly for commercialization. This approach will require additional effort to demonstrate comparability between the early-phase and commercial product, which potentially creates a risk of the need for additional clinical trials. An alternative approach is to develop a “commercializable” process early and leverage that process for all clinical development. This approach has the potential advantage of lowering risk to product comparability assessment, but requires additional early process development investment. The strategy presented in this paper is based on the latter approach, where the process development objective is to develop a commercial production cell line for first human dose (FHD) or first-in-human (FIH) application, and leveraging a “commercializable” platform for cell culture and downstream process development. At the heart of this strategy is the view that the production cell line is the foundation of any bioprocess, and thus, appropriate genetic characterization of the production cell line is absolutely crucial to the success of process development.
Phase-appropriate strategy for characterization of transgenes expressed in CHO cellsThe CMC development phases discussed in this manuscript refer to the three phases commonly associated with clinical and commercial development. The early phase begins from generation of the clonally derived production cell line and ends with the release of the formulated drug product for FHD/FIH. The mid-phase involves activities including product resupply for ongoing clinical trials (first efficacy dose [FED]) and development of the commercial manufacturing process. Late-phase development starts from the manufacture of product for the first registration dose (FRD) using commercial manufacturing processes and ends with process validation and submission of the product licensing applications. Table I summarizes the CMC development phase-appropriate activities for the characterization of the transgenes encoding the therapeutic proteins expressed in CHO cells.
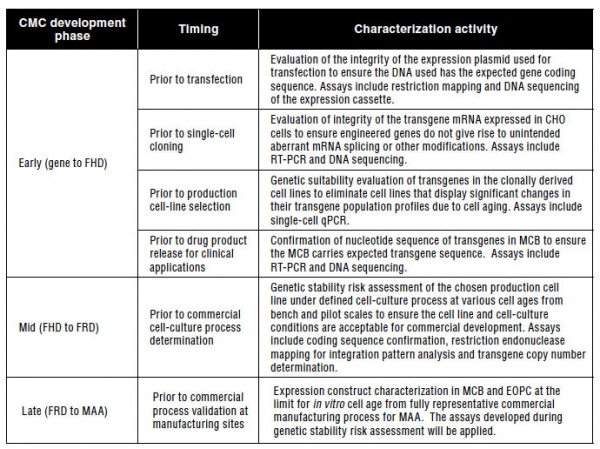 Table I: Phase-appropriate characterization activities for therapeutic transgenes expressed in Chinese hamster ovary (CHO) cells. FHD=first human dose, FRD=first registration dose, PCR=polymerase chain reaction, qPCR=quantitative polymerase chain reaction, MCB=master cell bank, RT-PCR=reverse transcription polymerase chain reaction, EOPC=end-of-production cells, MAA=marketing authorization application.
Table I: Phase-appropriate characterization activities for therapeutic transgenes expressed in Chinese hamster ovary (CHO) cells. FHD=first human dose, FRD=first registration dose, PCR=polymerase chain reaction, qPCR=quantitative polymerase chain reaction, MCB=master cell bank, RT-PCR=reverse transcription polymerase chain reaction, EOPC=end-of-production cells, MAA=marketing authorization application.
For clarity, two terms, genetic suitability and genetic stability, are used to describe the characterization of the transgenes expressed in CHO cells. The genetic suitability of a production cell line refers to the acceptability or appropriateness of the cell line for clinical drug substance manufacturing. In contrast to genetic suitability, more traditional genetic stability studies refer to the characterization of the production cell lines used for commercial drug substance manufacturing followed the ICH Q5B guidance. In conjunction with protein characterization and quality assessment, the genetic stability studies using nucleic acid techniques examine the integrity and stability of the expression construct, which could potentially impact protein integrity and process consistency. The genetic stability studies are performed in two phases: the mid-phase study, referred to as the “genetic stability risk assessment,” which evaluates the integrity and stability of the expression construct under defined commercial cell-culture process, and the late-phase study, referred to as the “expression construct characterization,” which generates the genetic characterization data package (based on the commercial cell-culture process) that forms part of the marketing authorization application (MAA).
Transgene characterization activities during early-phase development
As shown in Table I, four genetic characterization activities are implemented during early-phase development to minimize the risk of choosing inappropriate production cell lines. Those activities are summarized in the following passages.
Evaluation of the expression plasmid used for transfection
The cloning of genes coding for therapeutic proteins within an expression plasmid backbone is usually a straightforward procedure. It is a common practice and a recommendation of ICH Q5B to confirm the nucleotide sequence of the coding region of the gene(s) of interest and associated flanking regions that are inserted into the plasmid backbone. After the sequence of the expression plasmid is confirmed, the expression plasmid DNA is usually isolated at larger scale to prepare sufficient plasmid DNA to enable cell-line generation. From time to time, researchers encounter plasmid instability, such as loss of plasmid or changes in plasmid structure during large-scale bacterial cultivation (15). Through the evaluation of expression plasmid batches prior to transfection, the authors identified plasmid structural changes and point mutations that resulted from a larger-scale preparation, even though the expression plasmid was previously confirmed from a small-scale preparation (mini-prep). Because the issues were discovered and corrected prior to transfection, they did not cause significant delays in the generation of the production cell line.
Evaluation of integrity of the transgene mRNA expressed in CHO cells
RNA splicing is a natural process in mammalian cells that removes introns and joins exons in a primary transcript to create mature messenger RNA (mRNA) for translation. For biopharmaceutical development, the recombinant genes coding for the mature mRNA transcripts (i.e., cDNA versions) are commonly used for the production of drug substance to avoid complexity of RNA splicing in the production host. However, with the implementation of SAR studies and advancements in DNA manipulation techniques, more and more recombinant genes undergo extensive and complex genetic manipulation to improve the encoded candidate’s therapeutic traits. Although the protein engineering process has been successful in that regard, the full impact of some nucleotide sequence modifications is difficult to predict. It has been observed that several engineered antibody genes expressed in CHO cells -produced unintended transcripts—resulting from aberrant RNA splicing at cryptic splicing sites—in addition to the expected full-length transcript (2, 16). Aberrant mRNA splicing can lead to unexpected low-level expression of the recombinant transgenes (2) and/or give rise to truncated product-related impurities (16). In one case, the authors experienced an antibody-producing cell line, which had cryptic aberrant mRNA splicing that gave rise to a truncated light chain (LC) product. Although the truncated LC was effectively removed through the downstream purification process, the overall purification yield was significantly reduced (to approximately 10%). A new production cell line was subsequently generated for Phase II/III and commercial applications. The aberrant splicing was eliminated in the new cell line by site-specific mutagenesis at the cryptic splicing sites.
To mitigate the risk of unintended splicing, the authors identified and eliminated cryptic aberrant splicing proactively during clinical candidate selection to avoid potential delays to the clinical development or the need to switch cell lines for commercial applications. The authors’ experiences and the literature indicated that several publicly available splice-site-recognition programs were unable to identify the aberrant splicing sites used in the CHO cell environment (2). The most effective methods to identify the presence of aberrant splicing events are reverse transcription polymerase chain reaction(RT–PCR) and Northern blot. The authors’ data indicate that RT–PCR is a more sensitive method to detect low level of aberrant splicing compared with Northern blot (unpublished data). The potential bias due to PCR primer-binding capability can be overcome by applying multiple pairs of primers.
RT–PCR-based methods have been developed and implemented to screen all new clinical candidates for the aberrant splicing events in both transiently and stably transfected CHO cells before they are chosen to generate production cell lines supporting clinical development. This early screening approach has identified cryptic aberrant splice sites in multiple therapeutic candidates including monoclonal antibodies, bispecific antibodies, and fusion proteins, therefore preventing significant investment in the development of certain candidate-encoding genes (unpublished data).
Evaluation of transgene distribution profiles in CHO cell lines
The inherent adaptive ability of CHO cells and their capacity for the expression and secretion of recombinant proteins have been the most important factors that have enabled the adoption of CHO cells as the industry’s predominant host for development and manufacturing of therapeutic proteins (17). However, as immortalized cells, CHO cell lines exhibit a high level of genetic and phenotypic diversity and instability (17). In spite of repeated rounds of single-cell cloning by limiting dilution or florescence-activated cell sorting (FACS), clonally-derived CHO cell lines have often been observed to diverge, becoming a heterogeneous population over long periods of sub-culturing (17–22). Extensive efforts have been devoted to the screening process to identify desired characteristics and suitability of recombinant CHO cell lines.
Historically, suitability has been evaluated utilizing phenotypic measures (productivity) assessing cell-line productivity across a generational span encompassing the manufacturing window. Given the variability of the phenotypic assessment, the current and preferred measure of suitability is to assess the genetic profiles of the production cell-line population, thus obtaining a measure of the genetic consistency of the cell line across generations. The profiles provide an indication of significant loss of the transgene (% negative cells) or significant levels of genetic heterogeneity within the cell-line population (e.g., broad transgene copy distribution or standard deviation) across generations (12). Cell lines displaying genetic heterogeneity may have a higher risk of not meeting commercial manufacturing requirements and, therefore, should be eliminated during production cell-line screening. The methodology used for testing transgene heterogeneity was initially developed and implemented to evaluate the genetic suitability of clonally-derived CHO cell lines expressing IgG1 and IgG4 monoclonal antibodies. It has now been adapted to permit the evaluation of candidate cell lines expressing Fab, Fc fusion proteins, proteins (with or without glycosylation), as well as bispecific or bifunctional molecules. Undesired cell lines—measured by three parameters including the percentage of negative cells, standard deviation of cycle threshold (Ct) value (a measure of population heterogeneity), and mean Ct changes during aging (a measure of population drift) (12)—were identified regardless of the type of transgenes being used. In the authors’ experience, approximately 20% of clonally-derived candidate production cell lines analyzed showed significant transgene population heterogeneity over generations. As examples, Figure 1 shows the transgene distribution profiles of two cell lines expressing identical recombinant therapeutic proteins obtained from the same transfection. The cell line in Figure 1A (designated 3E4) represents a relatively homogenous transgene population across the generational span needed for the manufacturing process, while the cell line in Figure 1B (designated 7H2) displayed a significant population shift when the cells were aged, as indicated by the Ct mean change of >1 and overall large standard deviations across generations.
 Figure 1: Transgene copy number distribution profiles of cell lines expressing recombinant protein at generations of 0, 30, 45, and 60. Generation 0 (G0) represents the generation of a master cell bank, G30 represents the generation of cells harvested from a 5000-L bioreactor, and G60 represents the limit for in vitro cell age designed for a commercial manufacturing process. The distribution profiles shown in the scatterplot and histograms (1A: cell line 3E4; 1B: cell line 7H2) were generated by Oneway platform of JMP software (SAS Institute, Inc, Cary, NC) as outlined in (12). The cycle threshold (Ct) values of transgene were generated by a single-cell quantitative polymerase chain reaction (qPCR) assay. The number of tested single cells (number), mean and standard deviation (std dev) of the Ct values are shown below the plots.
Figure 1: Transgene copy number distribution profiles of cell lines expressing recombinant protein at generations of 0, 30, 45, and 60. Generation 0 (G0) represents the generation of a master cell bank, G30 represents the generation of cells harvested from a 5000-L bioreactor, and G60 represents the limit for in vitro cell age designed for a commercial manufacturing process. The distribution profiles shown in the scatterplot and histograms (1A: cell line 3E4; 1B: cell line 7H2) were generated by Oneway platform of JMP software (SAS Institute, Inc, Cary, NC) as outlined in (12). The cycle threshold (Ct) values of transgene were generated by a single-cell quantitative polymerase chain reaction (qPCR) assay. The number of tested single cells (number), mean and standard deviation (std dev) of the Ct values are shown below the plots.
Confirmation of nucleotide sequence of transgenes in the master cell bank
Although it has been a common practice to verify the nucleotide sequence of transgenes encoding the therapeutic proteins in master cell banks (MCB) for the MAA as recommended by ICH Q5B guidance, it was only recently recommended by the European Medicines Agency (EMA) that the sequence of the coding region should be confirmed prior to the initiation of clinical trials (23).
For recombinant CHO cell lines, transgenes are integrated into CHO chromosome. The most common technique to verify the nucleic acid sequence encoding the product is sequencing of coding regions amplified by the polymerase chain reaction (PCR) from pooled cDNA isolated from the production cell line. The nucleic acid sequence of the predominant transgene transcripts should be identical—within the limits of detection of the methodology—to the expected sequence encoding for the protein.
In summary, the early-phase characterization activities focus on evaluating and minimizing the potential risks associated with a transgene’s integrity and consistency when expressed in CHO cells. By implementing these activities, manufacturers can ensure that potential issues are identified during cell-line generation and are prevented from posing risk to the development of the manufacturing process. It should be noted that the transgene expression system used for the generation of a production cell line may also impact its integrity and stability. Two of the most common expression systems leveraged for the production of therapeutic proteins in CHO cells are the dihydrofolate reductase (DHFR)-based methotrexate (MTX) selection system and the glutamine synthetase (GS)-based methionine sulfoximine (MSX) selection system. Observations have been reported of genomic DNA mutations in the cell lines utilizing the DHFR/MTX system (24), and the mutation rates measured by 6-thioguanine (6-TG) assay positively correlated with the MTX concentrations used to select the recombinant cell lines (3). Because multiple rounds of amplification are often applied using the DHFR/MTX system—which can result in as much as a 1000-fold increase in transgene copy number (25)—more detailed DNA and amino acid sequence analyses may be necessary to ensure consistent product quality (3). In contrast, the GS-MSX expression system typically does not require multiple rounds of amplification and, thus, usually results in relatively low transgene copy numbers. The authors’ internal data indicate that the average transgene copy number is approximately 5 in 62 clonally-derived production cell lines generated leveraging the Lonza GS expression system. The relatively low transgene copy number may reduce the risk of DNA alterations, although it does not eliminate the possibility of modification (26).
Transgene genetic stability risk assessment during mid-phase development
Although significant efforts are devoted to the identification of production cell lines with the fewest potential risks for commercialization, many of these studies are performed using cells grown in shake flasks or small bench-scale bioreactors under nonoptimized cell culture conditions. Therefore, it is crucial to further evaluate the integrity and stability of the transgenes using commercial cell-culture conditions during the mid-phase development. This approach can enable corrective actions to be taken to avoid costly changes later. It has been reported that unexpected genetic alterations were identified in the MCB, the manufacturing working cell bank (MWCB), the end-of-production cell bank (EPCB), and in production cells (27).
Details of this evaluation, termed the “genetic stability risk assessment” can be found in Table I. Mid-phase assessment focuses on evaluating the impact of cell age and cell-culture process using DNA and RNA isolated from the MCB or premaster research cell bank (pmRCB), end-of-production cells (EOPC) from a typical defined cell-culture process, and EOPC inoculated from the proposed limit for in vitro cell age. It includes assessment of integrity of predominant coding transcripts, consistency of integration patterns, and average transgene copy numbers for the aforementioned samples. The established assessment methods will be applied to characterize the expression construct needed for future MAA. The tested limit for in vitro cell age will be used to propose the commercial manufacturing operating space for marketing applications.
Characterization of the expression construct for MAA
The ultimate goal of genetic characterization of the production cell line is to demonstrate the integrity and stability of the expression construct carrying the transgenes. This includes demonstrating that these transgene-carrying constructs are stably maintained from the starting cell banks (MCB and/or WCB) to the EOPC at the limit for the in vitro cell age inoculated from a WCB and harvested from the commercial drug substance manufacturing process. This characterization—which confirms that the commercial cell culture process does not lead to unintended changes in the transgenes—is performed not only to meet regulatory expectations for the MAA, but also to provide assurance of safety and manufacturing consistency when coupled together with product characterization and an analytical control strategy. The characterization, focused on the integrity and consistency of the expression construct, consists of three aspects:
• Verification of protein coding sequence in production cells through the end of production. This is commonly accomplished by nucleotide sequence analysis of the transgene-specific PCR products amplified from pooled cDNA.
• Assessment of integration patterns, which provides insight into potential insertions and/or deletions of the expression construct. The most common methodology for this assessment is restriction endonuclease mapping analysis by Southern blot.
• Determination of average transgene copy number. The current common method is quantitative PCR (qPCR). Transgene-specific qPCR assays and a normalizer qPCR assay targeting a host genome region are usually applied. The characterization, focused on the integrity and consistency of the expression construct, consists of three aspects, which are methods established during the mid-phase “genetic risk assessment”.
The in vitro cell age is defined by ICH Q5B as “measure of time between thaw of the MCB vial(s) to harvest of the production vessel measured by elapsed chronological time in culture, by population doubling (PD) of the cells, or by passage level of the cells when sub-cultivated by a defined procedure for dilution of the culture” (14). In experience with the authors’ CHO cell lines, a typical cell age from a 5000-L production bioreactor is found to be approximately 30 PD including the cell age of a WCB, seed train, and bioreactor expansion and growth in the production vessel. Extra cell culture passages in the cell expansion stages are added to generate the cells at the limit for in vitro cell age to increase flexibility of manufacturing operation. The typical limit for in vitro cell age is in total approximately 45–60 PD, depending on the growth rate of each individual cell line and the specific cell-culture expansion process. The mid-phase risk assessment enables the design of limit for in vitro cell age for the commercial process.
The impact of a change in cell-culture process after the commercial process has been defined should be evaluated to determine if re-testing of the cells at the limit of in vitro cell age is necessary. Changes in cell-culture process scale and/or manufacturing site may not require re-testing of cells at the limit for in vitro cell age, provided the available data meet the predetermined acceptance criteria and were obtained using cells with sufficient in vitro cell age to cover the additional cell generations resulting from the increased scale and cell-culture performance. Changes in media components and growth conditions often result in changes in cell culture profiles, and one should consider re-testing the integrity and stability of the expression construct at the limit for in vitro cell age in those situations.
Although a two-tier cell-banking system (MCB and WCB) is commonly established for the commercial manufacturing of drug substance, it is considered that the characterization of the expression construct for each WCB is unnecessary if the following criteria are met:
• The MCB has been fully characterized and the expression construct is confirmed to be stable. If the characterization cannot be carried out on the MCB, it should be carried out on each WCB.
• The cell age of the current and the future WCB, as well as the EOPC derived from the future WCB, will be controlled within the previously approved limit for in vitro cell age.
By implementing a development-phase appropriate genetic characterization strategy, manufacturers can be assured that the integrity and stability of transgenes in all clonally derived CHO cell lines intended for drug substance commercial manufacturing meet regulatory requirements for MAA. Harnessing this strategy has facilitated the early identification of unsuitable/unstable cell lines, thus enabling effective investments of time and resources only on those cell lines that are appropriate candidates. This strategy also meets business needs consistent with aggressive development timelines and the industry-wide movement toward more efficient practices, from therapeutic candidate selection to product launch.
Potential applications of new technologies for the characterization of transgenes
As the biotechnology industry continues to mature, so does the ability to understand the processes used to manufacture biopharmaceutical products. Part of this understanding involves the recognition and ability to characterize production cell lines as populations of cells exhibiting various levels of genetic and phenotypic heterogeneity. The described strategy includes characterization of production cell-line populations in addition to more traditional genetic characterization methodologies. Although this strategy has historically served well in accomplishing its intended purpose, it is also recognized that there is a need to continue to monitor and assess the potential value of new nucleic acid-based technologies, such as next-generation sequencing (NGS), to potentially further enhance the capability for genetic characterization (28). NGS can be utilized as a complementary/orthogonal analysis tool for the investigation of bioproduct-related impurities. NGS can also provide a means of better understanding impurities if they are related to genomic mutations or nutrient limitations in a cell-culture process. New approaches and technologies are becoming available and hold promise for permitting better characterization of cell lines and cell-culture processes, which could lead to improved insight into how to develop manufacturing processes more holistically. Implemented together, the described phase-appropriate genetic characterization strategy, orthogonal product characterization, and appropriate product control strategies will ensure the safety and efficacy of clinically validated therapeutic proteins.
Conclusion
Potential risks related to the integrity and stability of recombinant transgenes expressed in CHO cells are inevitable during biopharmaceutical development. These risks include unintended aberrant mRNA splicing in the coding regions due to the bioengineering process and potential instability of the integrated recombinant genes as a result of the global genetic instability inherent in CHO cells. The genetic characterization strategy described here can play a crucial role in assessing and managing these risks. The authors’ implementation of this strategy has prevented problematic expression construct/cell lines from being developed inappropriately, thereby enabling more efficient and effective process development programs. The data presented here demonstrate that the CHO production cell lines developed by the authors have relatively low integrated transgene copy numbers, and those genes are stably maintained from their respective master cell banks to the end of production cells at the limit for in vitro cell age used for drug substance manufacturing. To ensure the most effective genetic characterization strategy, new technologies will continue to be evaluated for potential future applications.
Acknowledgment
The authors would like to thank Christal Winterrowd and Monica Myers for providing copy number analysis and Dennis Gately and Matthew Hilton for providing technical input to the characterization strategy. We’d also like to thank Matthew Hilton, Michael De Felippis, Tongtong Wang, and Anli Ouyang for critical review of the manuscript and helpful discussions. In addition, the authors wish to recognize and thank Lonza Biologics (Slough, UK) for the licensing of and permission to modify the GS-CHO expression system.
References
1. J.M. Reichert, MAbs. 7 (1), pp. 1–8 (2015).
2. S.D. Wijesuriya, R.L. Cotter, and A.H. Horwitz, Protein Expr. Purif. 92 (1), pp. 14–20 (2013).
3. D. Guo et al., Biotechnol. Bioeng. 107 (1), pp. 163–171 (2010).
4. P. Zhang et al., MAbs 4 (6), pp. 694–700 (2012).
5. L. Feeney et al., Biotechnol. Bioeng. 110 (4), pp. 1087–1097 (2013).
6. Y. Yang et al., MAbs 2 (3), pp. 285–298 (2010).
7. D. Ren et al., J. Chromatogr. B, 879 (27), pp. 2877–2884 (2011).
8. ICH, Q6B, Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products, Step 4 version (1999).
9. S.L. Davies et al., Biotechnol. Bioeng. 110 (1), pp. 260–274 (2013).
10. L.A. Bailey et al., Biotechnol. Bioeng. 109 (8), pp. 2093–2103 (2012).
11. V. Paredes et al., Biotechnol. Lett. 35 (7), pp. 987–993 (2013).
12. L. He et al., Biotechnol. Bioeng. 109 (7), pp. 1713–1722 (2012).
13. R.A Heller-Harrison et al., “Advances in Protein Production” supplement to BioPharm Int. 22, pp. s16–27 (June 2009).
14. ICH Q5B, Quality of Biotechnological Products: Analysis of the Expression Construct in Cells used for Production of R-DNA Derived Protein Products, Step 4 version (1995).
15. F.A. Al-Allaf et al., 3 Biotech. 3 (1), pp. 61–70 (2013).
16. J.M. Reichert, N.M. Jacob, and A. Amanullah, Biotechnol. Prog. 25 (4), pp. 1198–1215 (2009).
17. F. Wurm, Processes, 1 (3), pp. 296–311 (2013).
18. M. Derouazi et al., Biochem. Biophy. Res. Communications 340 (4), pp. 1069–1077 (2006).
19. L.M. Barnes, C.M. Bentley, and A.J. Dickson, Cytotechnology, 32 (2), pp. 109–123 (2000).
20. N.S. Kim, S.J. Kim, and G.M. Lee, Biotechnol. Bioeng. 60 (6), pp. 679–688 (1998).
21. P.M. O'Callaghan and D.C. James, Brief Funct. Genomic Proteomic, 7 (2), pp. 95–110 (2008).
22. M. Kim et al., Biotechnol. Bioeng. 108 (10), pp. 2434–2446 (2011).
23. EMA/CHMP/BWP/534898/2008 Committee for Medicinal Products for Human Use (CHMP), Guideline on the requirements for quality documentation concerning biological investigational medicinal products in clinical trials (March 15, 2012).
24. R.J. Harris et al., Biotechnol. (NY) 11 (11), pp. 1293–1297 (1993).
25. R.J. Kaufman and P.A. Sharp, J. Mol Biol. 159 (4), pp. 601–621(1982).
26. H. Dorai et al., BioProcess Int. 5 (8), pp. 66–72 (2007).
27. J. Fu et al., J. Chromatog. B Analyt. Technol. Biomed. Life Sci. 908 (1), pp. 1–8 (2012).
28. S. Zhang et al., Biotechnol. Prog. 31 (4), pp. 1077–1085 (2015).
About the Authors
Luhong He, PhD, is senior research scientist, and Christopher Frye is research advisor, both in the department of Bioprocess Research and Development at Eli Lilly and Company. Email: [email protected].