The author discusses the current best practices in technical qualification of single-use systems.
By Weibing Ding, PhD
 Single-use technology has been around for approximately two decades if disposable capsule filters are taken into consideration. Aseptic connector devices, mixer bags, storage bags, and bioreactors followed. Currently, single-use systems (SUS) also include disconnectors, disposable filling needles, and sensors. The technology is still evolving, and the industry can expect further advances in the future.
Single-use technology has been around for approximately two decades if disposable capsule filters are taken into consideration. Aseptic connector devices, mixer bags, storage bags, and bioreactors followed. Currently, single-use systems (SUS) also include disconnectors, disposable filling needles, and sensors. The technology is still evolving, and the industry can expect further advances in the future.
The dynamic nature of SUS provides excitement for professionals from both suppliers and end users, who predominantly believe that SUS is a key technology for the future of biomanufacturing because of the following major advantages:
• Flexibility
• Less up-front capital investment
• No batch-to-batch cross contamination
• No re-use cleaning validation
• Less work on scale up
• Shortened time-to-patient.
When end users actually implement SUS in cGMP biomanufacturing processes, however, there are still many challenges including the following:
• Lack of thorough understanding of new materials (e.g., compatibility with processing fluids/extractables)
• Lack of deep knowledge of interaction between SUS and biologics
• Lack of good practices to prevent leakage of SUS (manufacturing/ shipping/handling)
• Limited interchangeability of components (e.g., connectors)
• Limited industrial and regulatory guidance on qualification of SUS.
These challenges have prompted both suppliers and end users to work collaboratively to adequately qualify SUS prior to its implementation.
SUS Process Development
Single-use systems are being used in almost all stages of biomanufacturing processes, including upstream, downstream, and fill/finish operations. Figure 1 depicts a typical monoclonal antibody (mAb) drug manufacturing process. Single-use bioreactors are widely used in cell line expansion, and the use at production scale is increasing, notably in contract manufacturing. Single-use buffer filtration system and storage bag application is common in downstream processes. Final drug-product sterile filtration and transfer using SUS has also being increasingly applied to fill/finish operations.
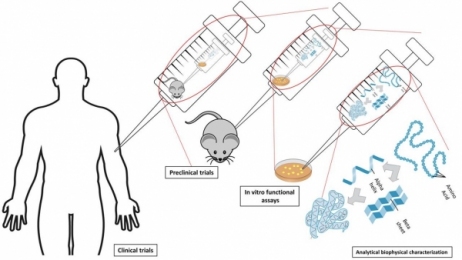 Figure 1: Typical mAb manufacturing process.
Figure 1: Typical mAb manufacturing process.
The implementation of SUS usually starts with development of the user requirement specification (URS). Typically, the development engineers at biopharmaceutical companies then work closely with the design engineers and application engineers from suppliers to select components and standard assemblies as available, or if not, generate suitable drawings, which will then be subjected to internal approval within the end-user company. There are many factors to be evaluated, including technical assessment, supply chain, business aspects, and supplier quality. PDA
Technical Report No. 66, Application of Single-Use Systems in Pharmaceutical Manufacturing, has been developed to provide comprehensive, high-level guidance about qualification of SUS (1). This paper focuses on technical assessment in more detail.
Technical Considerations: Chemical Compatibility
Unlike traditional stainless-steel equipment, SUS is generally made from polymers including plastics and elastomers. These polymers are obtained through polymerization of organic monomers in the presence of catalysts. As a result, they are susceptible to attack by organic solvents as well as acids and bases; some, at high concentrations, can even dissolve polymers. Because biomanufacturing may involve processing of fluids containing these chemical compounds, the chemical compatibility of single-use components with process fluids under process conditions should be the first factor to be evaluated.
Table I lists the common polymers used in the major single-use components. Table II provides the general chemical incompatibility of these polymers with common fluids. Because chemical compatibility is determined by the concentration of contacting fluid, the nature of the polymer, the contact time, and temperature, Table II can only be used as a general guideline. When one intends to use SUS with these fluids, a pre-use qualification test should be performed to validate that the process conditions are not incompatible with the material in question (2–4). The consequences of incompatibility would be leaks from the closed system (e.g., tubing, connector, chromatography column, and biobag), inaccurate readings from sensors, failed filtration (filter), and potential failures in process performance and product quality.
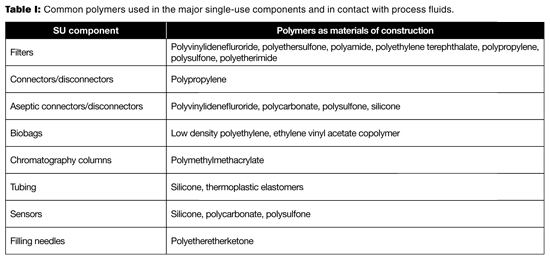 Table I: Common polymers used in the major single-use components and in contact with process fluids.
Table I: Common polymers used in the major single-use components and in contact with process fluids.
 Table II: General chemical incompatibility of these polymers with common fluids in bioprocessing* (see references 2-4).
Table II: General chemical incompatibility of these polymers with common fluids in bioprocessing* (see references 2-4).
In addition, pretreatment methods such as steam sterilization, e-beam irradiation, gamma irradiation, vaporized hydrogen peroxide, and ethylene oxide could have an effect on the properties of the polymers. Therefore, the specific process parameters of these pre-treatments should not exceed the supplier’s product claim, which should be proven by the supplier’s validation study.
Technical considerations: Fitness for purpose
SUS used for each unit operation should be fit for purpose. For example, a single-use bioreactor should be sterile and not adversely affect cell growth due to certain leachables. It should provide sufficient mixing and have acceptable gas exchange, and should not leak during operation. For buffer filtration and storage, the SUS should be sterile, not present any concerns for extractables/leachables, and not leak. For the SUS in fill and finish, it should be sterile, not present any concerns for extractables/leachables, not leak, and in addition, not raise any concerns for particulates and endotoxins.
The questions are: How can we ensure these requirements? Which part is the responsibility of the supplier? Which part is the responsibility of the end user?
In general, process validation remains the responsibility of the biopharmaceutical manufacturer. Supplier data should be appropriately used. Leveraging supplier data requires the end user to understand how the supplier data were developed and assess their suitability to the processes.
Regulatory Requirements
For finished pharmaceuticals, FDA has issued regulations explaining cGMPs in 21 Code of Federal Regulations (CFR) 210 and 211 (5, 6). For APIs or drug substances, however, the agency has not issued regulations. Nevertheless, the agency issues guidance documents to articulate its current practice on related topics. Although there is no specific regulation dedicated to SUS, FDA’s Q7A Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients (7), which is based on ICH Q7 (8), is a guidance document that should be studied, understood, and followed. Components and systems used for single-use technologies should be assessed for incoming testing. Key factors listed in the guidance document (7) include:
• “A supplier’s certificate of analysis can be used in place of performing other tests, provided that the manufacturer has a system in place to evaluate suppliers.
• “Supplier approval should include an evaluation that provides adequate evidence (e.g., past quality history) that the manufacturer can consistently provide material meeting specifications.
• “Processing aids, hazardous or highly toxic raw materials, other special materials, or materials transferred to another unit within the company’s control do not need to be tested if the manufacturer’s certificate of analysis is obtained, showing that these raw materials conform to established specifications. Visual examination of containers, labels, and recording of batch numbers should help in establishing the identity of these materials. The lack of on-site testing for these materials should be justified and documented.
• “Equipment should be constructed so that surfaces that contact raw materials, intermediates, or APIs do not alter the quality of the intermediates and APIs beyond the official or other established specifications.”
Failure to follow the guidance could result in receiving an inspectional observation from regulatory authorities (9).
Supplier Practices
The basic document generated by the supplier and shared with the end user is the validation guide for a component or a SUS. At a minimum, the following topics should be evaluated; for some components, other specific tests may also need to be performed:
• Materials of construction (Animal-derived ingredients free or meeting the EMA410.01 rev.03)
• Particulates/Endotoxin (United States Pharmacopeia [USP] <788>/USP <85>)
• Extractables (USP <661> and / or other more robust test designs [10])
• Biocompatibility (International Organization for Standardization (ISO) 10993-4)
• Cytotoxicity/Bioreactivity (USP <87>/USP<88>)
• Sterility (USP <71>)
• Transportation validation (ISTA-2A or American Society for Testing and Materials [ASTM] D7386-12)
• Shelf life (at least two years post gamma irradiation)
• Sterilization (ISO11137)
• Integrity or gross leak
• Functional tests.
Functional tests are specific to each SU component. For bioreactor bags, film gas barrier tests and film mechanical strength tests should be performed. Carbon dioxide, oxygen, and water vapor transmission studies can follow ASTM F2476, ASTM D3985/ASTM F1307, and ASTM F1249/ISO 15106-3, respectively. Mechanic strength tests include tensile strength (ASTM D882/ISO 527), tear strength (ASTM D1004), seal strength (ASTM F2097/ISO 15747), brittleness (ASTM D1790), and dynamic mechanical properties (ASTM D5026). In addition, drop tests on bags are performed according to ASTM D4169-05. For SU assemblies, the joints between tubing and barbs should be validated to prevent potential leakage.
In addition to the validation guide, suppliers may have additional supporting documents, including but not limited to detailed extractables report, certificate of quality, and certificate of gamma doses.
End User Practices
Aside from qualifying a supplier through a quality audit, a technical due diligence visit is necessary to understand the supplier’s manufacturing process, sources of variation, and their control. The goal is to ensure that the supplier can provide SUS or components with high quality consistently, which do not negatively impact the end user’s biomanufacturing process and/or drug product quality and safety. To achieve this goal, technical transparency is essential. During the visit, the technical team members from both companies should review test designs, confirm test methods/results, review test reports, and understand the test results. When needed, sub-suppliers should also be involved.
The supplier’s release criteria and end-user’s receiving acceptance criteria should be aligned with each other. These include, but are not limited to, packaging integrity, appearance/identity, contamination, and extractables assessment. The alignment of criteria should include any variability in appearance, such as minor blemishes on the exterior of SUS arising from transport and handling, which do not impair the function of the assembly. When developing specifications, functional specifications should be taken into account to allow SUS or component interchangeability wherever feasible.
The next step is to perform a gap analysis in the context of the specific application. The major points to consider are as follows:
• Is the extractables study relevant and report useful?
• Are the particulate/endotoxin tests adequate?
• Is the irradiation process validated and results certified?
• Is cell culture growth in the bioreactor acceptable?
• Is a gross leak test carried out prior to irradiation?
• Is shipping validation performed after irradiation?
• Does the use of the SUS affect product stability?
Based on the gap analysis, additional qualification tests/assessments may be warranted.
Best Practices
To ensure successful biomanufacturing, the SUS must not leak and must not adversely affect process performance and product quality during production. To ensure the drug product quality and patient safety, the SUS shall not be reactive, additive, or absorptive so as to alter the safety, identity, strength, quality, or purity of the drug product (11). The technical due diligence should be carried out prior to the implementation of SUS. Table III provides a partial list of best practices to consider in the technical qualification of SUS used in biomanufacturing (12–15).
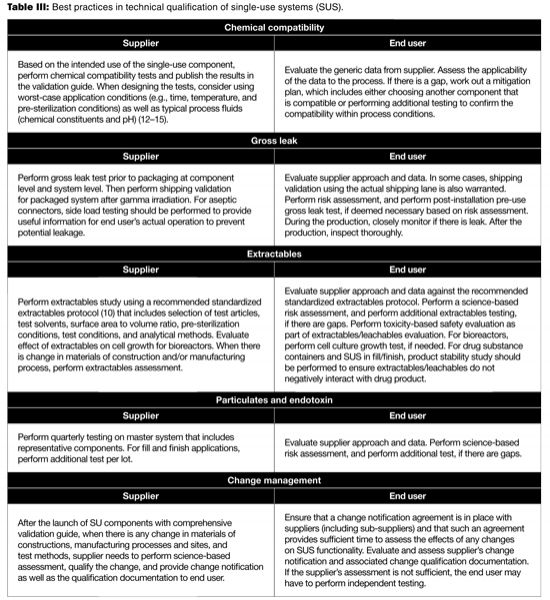 Table III: Best practices in technical qualification of single-use systems (SUS).
Table III: Best practices in technical qualification of single-use systems (SUS).
Conclusion
The partnership between end user and supplier is essential to successfully implement SUS in manufacturing drugs to achieve success in implementation and continued operation, and to ensure the quality and safety of the drug products. The supplier needs to be fully transparent about materials of construction, supply chain, manufacturing process, sources and control of variability, impact of changes, and qualification/validation. The end user needs to perform quality audits and technical due diligence visits to ensure compliance as well as a deep understanding of all these aspects, and then carry out science-based risk assessment considering the proximity of SUS to the final drug product container closure system by applying a phase-appropriate approach. Any gaps found should be bridged or filled in to ensure the successful implementation of SUS in biomanufacturing.
Acknowledgments
The author greatly appreciates Duncan Low’s, Executive Director, Process Development, Amgen, guidance and constructive review of the manuscript. In addition, the author sincerely thanks the collaboration from Amgen’s Raw Materials & Devices, Supplier Quality Management, Global Strategic Sourcing, and Process Development, as well as major suppliers for SUS.
About the Author
Weibing Ding, PhD, is principal scientist Process Development, Amgen Inc., Thousand Oaks, CA 91320.
ALL FIGURES ARE COURTESY OF THE AUTHOR
References
1. PDA, Technical Report No. 66, Application of Single-Use Systems in Pharmaceutical Manufacturing (PDA, 2014).
2. H. Domininghaus, Plastics for Engineers, Materials, Preperties, Applications (Hanser Publishers, 1993).
3. Plastics Design Library, Chemical Resistance, Second Edition (William Andrew Inc., April 1996).
4. P. A. Schweitzer, Corrosion Resistance Tables, Third Edition, Revised and Expanded (Marcel Dekker, Inc., 1991).
5. FDA, Part 210 Current Good Manufacturing Practice in Manufacturing, Processing, Packing, or Holding of Drugs; General.
6. FDA, Part 211 Current Good Manufacturing Practice for Finished Pharmaceuticals.
7. FDA, Guidance for Industry, Q7A Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients.
8. ICH, Harmonised Tripartite Guideline, Q7 Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients
9. FDA, Form 483, 2013.
10. W. Ding et al., Pharmaceutical Engineering (November/December 2014), pp74-85
11. FDA, CGMP for Finished Pharmaceuticals; 21 CFR 211.65(a)
12. V. Vinci and S Parekh, Handbook of Industrial Cell Culture: Mammalian, Microbial and Plant Cells (Humana Press, 2003).
13. P. Culter, Protein Purification Protocols, Second Edition (Humana Press, 2004).
14. F. Jameel and S. Hershenson, Formulation and Process Development Strategies for Manufacturing Biopharmaceuticals (Wiley, 2010).
15. L. Ducry, Antibody-Drug Conjugates (Humana Press, 2013).