An approach for establishing the CQAs of a mAb product by evaluating impact and uncertainty during risk assessment.
 A critical quality attribute (CQA) has been defined as “a physical, chemical, biological, or microbiological property or characteristic that should be within an appropriate limit, range, or distribution to ensure the desired product quality” (1). Biotech therapeutics, particularly complex products such as monoclonal antibodies (mAbs), can have numerous quality attributes that can potentially impact safety and/or efficacy of the product (2). Identifying CQAs for a biotech therapeutic is the first and arguably the most difficult step in implementation of quality by design (QbD) for development and production of biopharmaceuticals (3, 4).
A critical quality attribute (CQA) has been defined as “a physical, chemical, biological, or microbiological property or characteristic that should be within an appropriate limit, range, or distribution to ensure the desired product quality” (1). Biotech therapeutics, particularly complex products such as monoclonal antibodies (mAbs), can have numerous quality attributes that can potentially impact safety and/or efficacy of the product (2). Identifying CQAs for a biotech therapeutic is the first and arguably the most difficult step in implementation of quality by design (QbD) for development and production of biopharmaceuticals (3, 4).
Even if a firm chooses not to pursue intensive studies to map out complete design spaces for their manufacturing process, and instead opts for what the International Conference on Harmonization (ICH) Guideline Q11 refers to as a “traditional approach,” the ability to differentiate between what is and is not important for a molecule can drive sound decision-making in process development, which can lead to improved efficiency, cost savings, and more consistent product quality (5). And for those sponsors who do embrace a more comprehensive, “enhanced approach” to development, a solid understanding of product quality attributes serves as the touchstone upon which process design and integrated control strategies are built.
Structural characterization is used to assess the CQAs of biopharmaceutical products. The structural data must be supported by functional data to establish a structure-function relationship. In turn, these data can then be used to define the structural components’ impact on the activity of the product. Furthermore, the characterization data obtained are essential for product development and regulatory acceptance. Characterization of multiple product batches is essential to demonstrate to the regulatory body that the manufacturer has control of the manufacturing process. This is achieved by analyzing a number of batches of product and comparing the data. Significant differences between batches need to be investigated and their impact on the function of the product assessed. This comparison in the QbD paradigm also centers around the CQAs.
The European Medicines Agency’s guideline covering “Production and Quality Control of Monoclonal Antibodies” requests that “the mAb should be characterized thoroughly” (6). “This characterization should include the determination of physicochemical and immunochemical properties, biological activity, purity, impurities, and quantity of the mAb, in line with ICH Q6B guideline” (7). The EMA mAb guideline also draws attention to a number of structural features including N- and C-termini (in particular pyroglutamic acid at the N-terminus and lysine at the C-terminus of the heavy chain), free sulfhydryl and disulphide bridge structure, glycosylation (in particular the degree of mannosylation, galactosylation, fucosylation, and sialylation), and other post-translational modifications (e.g., deamidation, oxidation, isomerisation, fragmentation, and glycation).
In this 30th article of the “Elements of Biopharmaceutical Production” series, the authors focus on proposing an approach towards establishing CQAs for a mAb therapeutic product.
Product risk assessments
Identification of CQAs is often performed through a series of product risk assessments conducted over the program lifecycle, the first of which should be performed early in development to bring clarity to the goals of the Phase I process (4). Although the criticality of some attributes at this stage may still be somewhat based on speculation, these “potential CQAs” (pCQAs) serve as both a baseline for development to proceed and a gap analysis to identify which attributes would benefit from further in-vitro or in-vivo studies to ascertain their true impact on efficacy and safety. As the molecule progresses through development and more is learned about the relationship between product attributes and their impact (or non-impact) on potency, pharmacokinetics (PK), or safety, these pCQAs can be further refined and accordingly designated as CQAs as the product approaches the licensure application.
Because assigning criticality to an attribute hinges upon the question of risk posed to product safety and efficacy, a well-rounded product risk-assessment team should include representatives with expertise in PK, toxicology, in-vivo biology, and clinical management. A risk-ranking and filtering approach developed on the firm’s experiences and regulatory feedback may be used. Many such tools have been presented in the literature, and a firm can pick a tool of their choice as long as the basis is rational and it has been justified that the tools are used consistently (8, 9). The risk-assessment team first compiles a list of all the quality attributes of the product and systematically evaluates each attribute with regards to two factors: impact and uncertainty.
For impact, the team determines the severity of the consequences that would be associated with failure to control the attribute. The team considers the effects of the attribute not only on potency with respect to the intended mechanism of action, but also PK, pharmacodynamics (PD), immunogenicity, off-target effects, and direct impact to safety. The data used in the impact assessment may come from structure-activity relationship (SAR) studies, nonclinical studies, clinical exposure history, and toxicology reports (4). For platform molecules, or new products with structural homology to established classes (e.g., Fc fusion proteins, pegylated proteins), the team can leverage information from related proteins. Conversely, the more novel the protein is, the less opportunity there may be to apply knowledge across products.
After assigning an impact score, the team then evaluates the quantity and relevance of the body of data used in its assessment and assigns an uncertainty score to the attribute. The team considers its degree of reliance on in-vitro vs. in-vivo data, the availability of molecule-specific data pertaining to potency and PK, the relevance of data leveraged from related molecules, and the range of clinical exposure. Process additives undergo a similar assessment, which focuses on the sufficiency of toxicology data and the additives’ history of use. In general, the assessment of product quality attributes for novel proteins will have a higher degree of uncertainty than platform molecules in early development. For these products, one should strongly consider conducting the initial product risk-assessment exercise early in the development cycle to align the organization on a commonly-recognized target product profile. Otherwise, cell culture, purification, and drug product development may put undue importance on meeting certain criteria that are ultimately not critical, resulting in suboptimal processes that make unnecessary trade-offs between attributes.
Once the impact and uncertainty scores have been assigned, the product of these two values constitutes the risk priority number (RPN) for the attribute (9). Although the scoring system may define a numerical threshold for which attributes would receive a CQA or pCQA designation, the degree of confidence in assigning the impact and uncertainty scores must be kept in mind to avoid over-interpretation of the analysis. Instead, the authors have found that viewing a product’s quality attributes from a more holistic view, using the scores to generally characterize their degree of criticality with labels such as “high,” or “moderate-to-low,” is preferable (4). This is also consistent with guidance from regulators, who encourage firms to view attributes as lying along a “continuum of criticality,” in which attributes warrant different degrees of control depending on how critical they are and how readily they can be controlled through the process. It should be highlighted that while an attribute can be “less critical”, it does not mean that a control strategy is silent with regards to its control; every attribute requires a control strategy commensurate with its degree of risk.
It is also important to note that within the context of a product risk assessment, it is generally a good practice to exclude process capability considerations and the extent to which they can mitigate the risk. The fact that a highly critical attribute is easily controlled through the process, even to the point of not requiring routine testing, should be captured separately in process risk assessments and in the overall control strategy design and justifications. By evaluating attribute criticality solely on the basis of impact and uncertainty, the product risk assessment only needs to be revised when new information is discovered regarding the biology or toxicity of the attributes themselves, and not every time a process change is made (8).
In general, the pCQAs identified in the initial product risk assessment fall into two categories:
• pCQAs that are known or are highly likely to directly impact safety or efficacy will ultimately become CQAs. Attributes such as residual host-cell proteins, endotoxin, protein aggregates, and biological potency, which may initially be assigned as pCQAs during Phase I development, are highly critical in such a fundamental way that no amount of additional experimental study will alter their assignment as CQAs later in development.
• pCQAs whose impact on efficacy is unknown or uncertain will likely be the main focus of CQA determination studies. These are attributes whose criticality can vary on a molecule-by-molecule or class-by-class basis, and therefore, can benefit most from further experimental studies to accurately define their impact to product performance. These attributes typically include post-translational modifications and stability-indicating chemical changes to the molecule, such as glycosylation, charge isoforms, phosphorylation, oxidation, and deamidation.
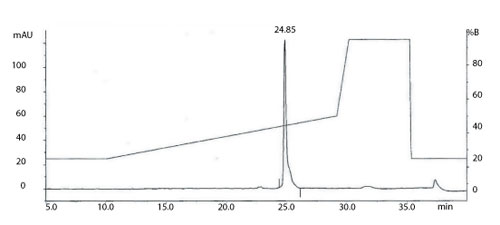
Figure 1: The total ion current (TIC) chromatogram obtained from on-line reverse phase-high-performance liquid chromatography with electrospray mass spectrometry (RP-HPLC/ES-MS) analysis of a monoclonal antibody. All figures are courtesy of the authors.
Mass Spectrometry for Molecular Weight Measurement and Peptide Mapping
Intact molecular weight measurement and peptide mapping are routinely used to assess and compare the protein backbone of the light and heavy chains of mAbs (10, 11) and can be valuable tools in determining product CQAs. Intact molecular weight analysis using high end quadrupole-time-of-flight (Q-TOF) mass spectrometers is able to produce mass accuracy within a few Daltons for intact mAbs. This information, coupled with analyses following reduction and de-N-glycosylation, allows an initial assessment of the intactness of the product and a number of the post translational modifications present on the molecule. Figure 1 shows the total ion current (TIC) chromatogram obtained from on-line reverse-phase, high-performance liquid chromatrography with electrospray mass spectrometry (RP-HPLC/ES-MS) analysis of a mAb.

Figure 2: Transformed electrospray mass spectrum prepared from m/z data acquired during elution of the main component observed during on-line liquid chromatography-mass spectrometry (LC-MS) analysis of a monoclonal antibody.
A transformed electrospray mass spectrum created from m/z data acquired during elution of the peak observed at 24.85 minutes is shown in Figure 2. These data combined with the data obtained from analysis of the mAb following reduction, and the heavy chain component following de-N-glycosylation (transformed mass spectra shown in Figures 3-5), allow the intactness of the mAb to be assessed. The data also provide an overview of the N-linked glycosylation, allow an assessment of the level of C-terminal Lysine (at the C-terminus of the heavy chain) and also suggest, for this particular product, that a portion of the heavy chain exists in glycated form. These analyses provide a significant amount of information from rapid and relatively straightforward experiments. The data obtained from analysis of the CQAs of further batches or reference materials in parallel can be used to provide an assessment of comparability in terms of batch to batch or indeed biosimilar to reference medicinal product.
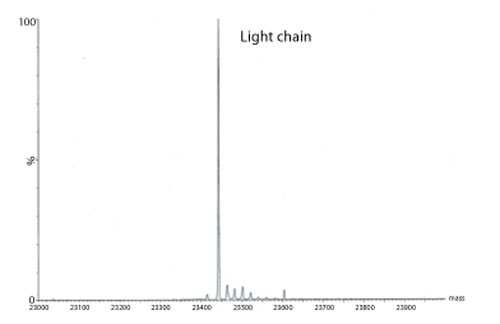
Figure 3: Transformed electrospray mass spectrum prepared from m/z data acquired during elution of the light chain during on-line liquid chromatography-mass spectrometry (LC-MS) analysis of a reduced monoclonal antibody.

Figure 4: Transformed electrospray mass spectrum prepared from m/z data acquired during elution of the heavy chain during on-line liquid chromatography-mass spectrometry (LC-MS) analysis of a reduced monoclonal antibody.
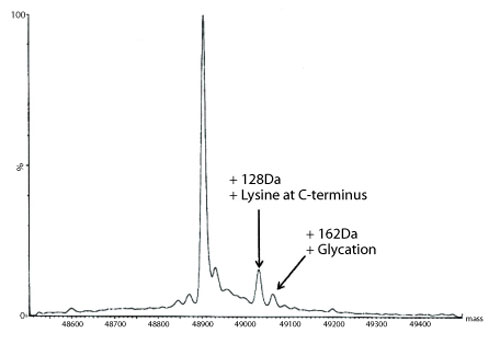
Figure 5: Transformed electrospray mass spectrum prepared from m/z data acquired during elution of the heavy chain during on-line liquid chromatography-mass spectrometry (LC-MS) analysis of a reduced de-N-glycosylated monoclonal antibody.
Peptide mapping is normally used to confirm these assignments and also allow further assessment of the protein backbone, particularly for post translational modifications. Figure 6 shows the basic process comprising reduction/alkylation, digestion, and enzymatic release of peptides (and glycopeptide) from a mAb for analysis.

Figure 6: Preparation of peptides for liquid chromatography-mass spectrometry (LC-MS) peptide mapping analysis.
The products of digestion are usually analysed by on-line LC-MS. The TIC data obtained from peptide mapping of two batches of mAb are shown in Figure 7 and allow a visual comparability assessment to be made.

Figure 7: On-line liquid chromatography-mass spectrometry (LC-MS) analysis of a reduced/alkylated monoclonal antibody following digestion
with trypsin.
Low energy (for peptide molecular weight) and high energy (for sequence assessment) mass spectrometric detection of the peptides during elution allow a further two dimensions of analysis. The data produced also allow a comparability assessment of the N- and C-termini of each chain (in particular presence and relative levels of pyroglutamic acid at the N-terminus and lysine at the C-terminus of the heavy chain), free sulfhydryl and disulphide bridge structure (from a comparison of reduced and non-reduced digests), glycosylation (in particular the degree of mannosylation, galactosylation, fucosylation, and sialylation), and other post-translational modifications (e.g., deamidation, oxidation, isomerization, fragmentation, and glycation).
Gaining Product Knowledge Through SAR and Pk Studies
For platform-driven molecules, such as mAbs, a pre-established set of pCQAs can often be applied across programs, but care must be taken to ensure that they are relevant to each new molecule under development.
For example, as part of early-stage product characterization, a new mAb was tested to determine whether its susceptibility to methionine oxidation was typical to that of other platform mAbs. Figure 8A illustrates the levels of site-specific methionine oxidation measured by LC-MS peptide map in the presence of increasing concentrations of an oxidizing agent. This study found that although the two conserved methionine residues in the Fc domain were prone to oxidation, the most susceptible methionine residue was located within the complementarity determining region (CDR). This finding prompted additional follow-up to determine the criticality of oxidation at the CDR site. Oxidized preparations of the mAb were prepared, characterized to ensure suitability of use (i.e., lack of extensive aggregation or fragmentation), and were then submitted for potency testing by binding assay. As shown in Figure 8B, preparations of the mAb containing up to 78% oxidized methionine at the CDR site were found to be equipotent to unstressed control material, providing assurance that this product quality attribute was not a critical determinant of product activity.

Figure 8: Structure-activity relationship study of complementarity determining region (CDR) methionine oxidation for a monoclonal antibody. (A) Site-specific oxidation susceptibility with increasing concentrations of hydrogen peroxide reveals a CDR methionine residue (red circles) that is most sensitive to modification, as determined by liquid chromatography-mass spectrometry (LC-MS) peptide mapping. (B) CDR binding activity assay, showing that oxidized preparations were equipotent to control.
When SAR studies are conducted with legacy programs, the longer history of product development can be turned to one’s advantage, especially if multiple cycles of process development can provide a valuable source of materials for study. For example, terminal galactosylation was identified as a pCQA for a mAb, largely due to a high degree of uncertainty regarding its impact on efficacy. Because the levels of biantennary had changed over the course of development, archived materials could be incorporated into SAR studies to investigate CDR binding and FcγRIIIa binding activities with the drug substance lots actually used in Phase 1 studies. With agalactosyl (G0F) glycan levels from 36-75% adequately covered by clinical materials, only a hypogalactosylated preparation of the mAb (via galactosidase digestion) was needed to help supplement the study. Figures 9A and 9B summarize the results of these studies. It was found that CDR binding activity was unaffected by galactosylation of the Fc glycan, while FcγRIIIa binding activity decreased modestly with increased G0F content, consistent with literature reports. On the basis of these data, as well as the extensive clinical history that was established and the wealth of relevant literature, Fc glycan galactosylation was deemed to be a moderately critical quality attribute for this molecule.

Figure 9: Studying the effects of Fc glycan galactosylation on binding activity of a monoclonal antibody. Potency testing across a wide range of agalactosyl glycan content finds no correlation between levels of G0F glycan and complementarity determining region (CDR) binding activity (A), but a modest decrease in FcγRIIIa binding activity was observed with increased agalactosyl levels (B).
Assessing the impact of an attribute on PK can be challenging if the attribute of interest is inherently microheterogeneous, such as glycosylation. But in some cases, powerful analytical tools such as mass spectrometry, can be employed to deconvolute the results of PK studies conducted with heterogeneous source materials. To study the effects of glycan structure on the clearance of an Fc receptor fusion protein, an affinity extraction method was developed that could specifically recover the drug from cynomolgus monkey serum samples collected at various timepoints. Quantitative mass spectrometry was then used to determine whether site-specific glycan profiles changed between timepoints, which would suggest preferential clearance of certain glycoforms. Figures 10A and 10B illustrate the results from this study, which found that the criticality of glycan structure was highly site-dependent. The composition of glycans at the Fc site (see Figure 10A) was essentially unchanged over 22 days, with percentages of G0F, G1F, and G2F (biantennary glycans with 0, 1, or 2 galactoses) holding steady for the duration of the study. By contrast, Figure 10B shows that the relative population of sialylated biantennary glycans on the receptor domain, such as A2G2F, increased slightly at the expense of the asialylated glycan G2F over time. This example underscores the importance of considering criticality for some attributes on a site-by-site basis; a given glycan (G2F) can be non-critical at the Fc, but moderately critical on the receptor.
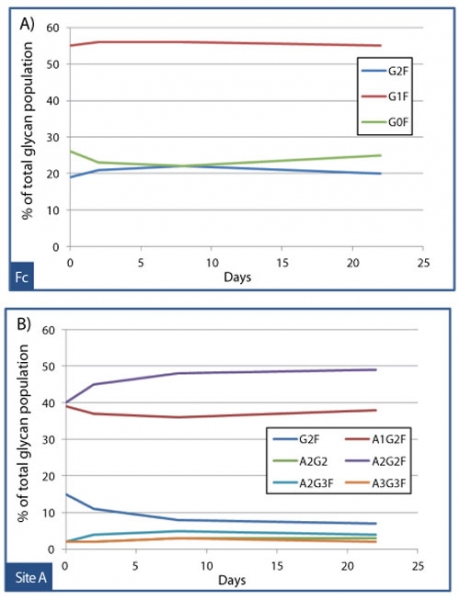
Figure 10: Affinity extraction and liquid chromatography-mass spectrometry (LC-MS) peptide map glycoprofiling to monitor site-specific dependent clearance of an Fc fusion protein. Over 22 days, the glycan population at the Fc site held steady (A), while a site on the receptor domain “site A” exhibited slight preferential clearance of the asialylated biantennary glycan, G2F (B).
Summary
Product risk assessment can be a valuable tool to not only help direct the course of process development and control strategy design, but also to identify opportunities to gain better understanding of what is truly important about the molecule itself. Designed experiments, using well-characterized test articles and advanced analytical methods, can help resolve uncertainties surrounding attribute criticality. Ultimately, a clear picture of attribute criticality, followed by mapping of their linkages to process parameters, can enable well-justified, science-based control strategies that put the right controls at the points where they can be most effective in ensuring product efficacy and patient safety.
Acknowledgments
The authors would like to thank Li Zang, Richard Strong, and Xiaoping Hronowski for their contributions to the SAR and PK studies cited in this publication.
References
1. ICH, ICH Harmonized Tripartite Guideline Q8: Pharmaceutical Development, Step 4 version (August 2009).
2. A. S. Rathore, Trends Biotechnol. 27 (12) 698-705 (2009).
3. A. S. Rathore and H. Winkle, Nature Biotechnol. 27 (1) 26-34 (2009).
4. A. S. Rathore, Trends Biotechnol. 27 (9) 546-553 (2009).
5. ICH, ICH Harmonized Tripartite Guideline Q11: Development and Manufacture of Drug Substances (Chemical Entities and Biotechnological/Biological Entities), Step 3 version (September 2011).
6. EMA, Guideline on Development, Production, Characterization and Specifications for Monoclonal Antibodies and Related Products, EMEA/CHMP/BWP/157653/2007 (December 2008).
7. ICH, ICH Topic Q6B Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products, Step 5 version (September 1999).
8. R.J. Seely and J. Haury, “Applications of Failure Modes and Effects Analysis to Biotechnology Manufacturing Processes” in Process Validation in Manufacturing of Biopharmaceuticals, A.S. Rathore and G. Sofer, Eds.) Taylor & Francis (2005) pp 13-30.
9. CMC Biotech Working Group, A-Mab: A Case Study in Bioprocess Development. October 2009, accessed June 5, 2014.
10. S. Kozlowski, “Implementation Activities for QbD: FDA Office of Biotechnology Products,” presentation at 2010 WCBP CMC Strategy Forum (Bethesda, MD, 2010).
11. L. Zang, X. L. Hronowski, Y. Lyubarskaya, A. Buko, H. Madden, W. Meier, R. Mhatre, “LC-ESI/MS and MALDI-MS for Monitoring of Glycoform-Related Clearance of a Complex Glycoprotein in Cynomolgus Monkeys,” presentation at the 58th ASMS Conference on Mass Spectrometry (Salt Lake City, UT, 2010).
About the Authors
Anurag S. Rathore is a professor at the Department of Chemical Engineering, Indian Institute of Technology, Hauz Khas, New Delhi, 110016, India, [email protected]; Andrew Weiskopf is director, Technical Development, Biogen Idec, 14 Cambridge Center, Cambridge, MA 02142; and Andrew J. Reason is UK business manager, Life Science Services, Group Manager SGS M-Scan Europe.