The authors outline cell-line development and process scale-up for an antibody program in which the antibody requires additional processing by a site-specific enzyme for correct functionality.
By Gregory Bleck, Dona York, David Rabuka
 The number of more complex biopharmaceuticals in development continues to increase. These include molecules such as virus-like particle (VLP) vaccines, proteins that require cleavage, or other additional post-translational modifications not typically performed by Chinese hamster ovary (CHO) cells, and more recently, numerous different multispecific antibody platforms. Each of these classes of molecules requires multiple proteins to be expressed at differing ratios to produce the correct biopharmaceutical. Many VLPs, for example, require two or more proteins to be produced in the right proportion to get the correct viral structure to mimic the virus itself.
The number of more complex biopharmaceuticals in development continues to increase. These include molecules such as virus-like particle (VLP) vaccines, proteins that require cleavage, or other additional post-translational modifications not typically performed by Chinese hamster ovary (CHO) cells, and more recently, numerous different multispecific antibody platforms. Each of these classes of molecules requires multiple proteins to be expressed at differing ratios to produce the correct biopharmaceutical. Many VLPs, for example, require two or more proteins to be produced in the right proportion to get the correct viral structure to mimic the virus itself.
In the case of a protein needing cleavage or another post-translational modification, the modifying protein needs to be produced at high enough levels to fully process all the biopharmaceutical at high titers without interfering with normal cell growth and the secretion of the molecule. Finally, some multispecific antibody platforms require three or more chains/subunits to be expressed at ratios that give the highest yield and the required structure for these molecules to be active.
Producing these multisubunit molecules or processing proteins in the correct proportions to obtain maximum titer and biopharmaceutical quality can be difficult using traditional cell-line development technologies. This is mainly due to problems getting different levels of protein expression for each of the multiple proteins needed for function and the ability of the system to obtain stable expression of each of those proteins over multiple generations in culture. In this article, the authors outline cell-line development and process scale-up for an antibody program in which the antibody requires additional processing by a site-specific enzyme for correct functionality. Cell lines and processes yielding titers of 2.5-5.0 g/L of highly processed antibodies were generated, resulting in specific productivities of 40-75 picograms/cell/day.
Characteristics of cell-line engineering technologies that aid in complex biopharmaceutical development programs
Stability (genetic and expression)
Expression of all proteins required to produce the complex molecule needs to be stable through the cell-line development, subsequent scale-up, and eventually, large-scale manufacturing. Because balancing ratios of individual proteins is so crucial for these types of programs, any shift in amount of each protein being produced during cell expansion and manufacturing can greatly impact the quality of the final product.
Ability to modulate ratios of protein expression
Different levels of protein expression are typically needed to maximize the production of a complex protein molecule. If a modifying enzyme is required in the process, a sufficient level of the enzyme is needed to fully modify the therapeutic protein; however, too much expression may affect cell growth or other cell behaviors. In the case of multispecific antibody platforms, certain unwanted chain pairings may occur more easily, prompting a desire to limit the production of that chain or subunit relative to the other ones being produced.
High gene insertion efficiency
The ability to efficiently insert genes into cells makes balancing ratios of protein expression a much easier process and shortens timelines. Complex protein molecules can require multiple cycles of gene insertion. Therefore, having a highly efficient gene insertion system resulting in a majority of cells containing the gene is important for both the protein product quality as well as shortening the project timeline.
Low-level use of selection markers
Many complex molecule programs rely on using numerous different gene constructs, each containing a different selectable marker. This use of multiple selectable markers in a cell line can complicate cell-culture processes, adding an increased burden on the cell-line machinery to produce multiple extra proteins. In addition, problems may arise during selection that affects the ability to identify clones that stably express the correct protein.
Protein conversion by FGE
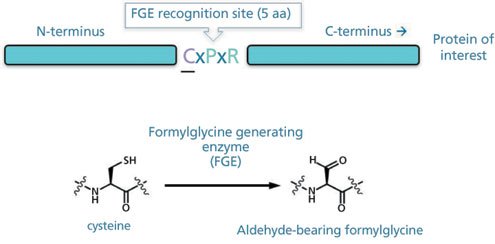 Figure 1: Formylglycine-generating enzyme (FGE) protein recognition site and conversion process in the context of a recombinant protein of interest.
Figure 1: Formylglycine-generating enzyme (FGE) protein recognition site and conversion process in the context of a recombinant protein of interest.
Formylglycine-generating enzyme (FGE) is an enzyme expressed in virtually all mammalian cells as well as most other eukaryotic and prokaryotic cell types (1). In the endoplasmic reticulum during protein synthesis, FGE performs the conversion of a cysteine (Cys) amino acid to a formylglycine (fGly) amino acid. This conversion requires FGE expression in the presence of the consensus sequence, cysteine-X-proline-X-arginine, where X represents any amino acid other than proline. FGE catalyzes the oxidation of the Cys thiol to an fGly aldehyde. In normal cells, sulfatase enzymes are substrates for FGE and they require the consensus sequence along with the Cys to fGly conversion in their active site to be functional. Most any recombinantly expressed protein can be engineered to contain the FGE consensus sequence at single or multiple locations. Subsequent conversion of the Cys to the fGly results in a chemical handle (the aldehyde) that can be used for site-specific chemical conjugation (Figure 1) (2, 3, 4, 5). CHO cells produce endogenous active FGE; however, there is not enough FGE to perform full conversion of the Cys to fGly in proteins expressed with the consensus sequence, especially when the engineered proteins are produced at high titers. Also recently, the FGE enzyme was shown to require copper (II) for full activity (6, 7), so supplementation of copper (II) sulfate to the media optimizes amino acid conversion.
Cell-line engineering for high-level protein expression and proper enzyme conversion
The goal of this case study was two-fold: to generate a stable CHO cell line expressing consistent high levels of FGE that could be used for a number of different programs, and to use the developed FGE cell line to produce cell lines and processes capable of expressing and converting high titers of tagged antibody. The FGE cell line needed to have all the normal phenotypic traits of the base CHO cells, but in addition produce enough FGE to allow full conversion of the tag at commercially viable titers and consistently do this over the number of cell generations needed for a commercial process. The subsequently tagged antibody-producing cell lines were required to consistently produce fully converted antibody at titers greater than 1 gram/L in a traditional fed-batch manufacturing process.
The GPEx technology (Catalent) was used for all of the cell-line engineering. The GPEx method is based on the use of replication-defective retroviral vectors (retrovectors) to actively insert the desired genes into the genome of dividing cells. By controlling the number of retrovector particles accessing the cell, multiple gene insertion can be achieved without any of the traditional amplification steps (8). This was important for this study to obtain enough FGE production in the context of high antibody production.
Development and selection of high-expressing FGE clonal cell line
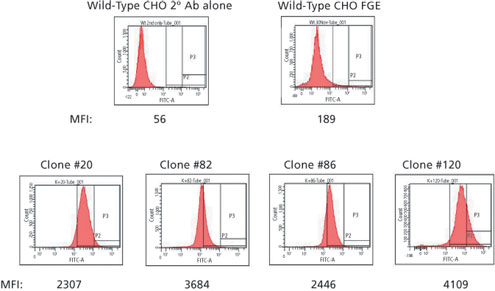
Figure 2: Flow cytometry analysis of wild-type Chinese hamster ovary (CHO) cells and clones overexpressing formylglycine- generating enzyme (FGE). Mean fluoresce intensity (MFI) examined for each cell line.
A master cell bank of our internal CHO cell line was the parent cell line for this work. The gene encoding human FGE was inserted into this line using the GPEx process. Two cycles of transduction were performed on the cells, and the resulting pool of cells was expanded for further development. Single-cell clones were isolated from the cell pool using limited dilution cloning. Approximately 120 clonal lines were initially screened. At the time, a high-throughput detection method for FGE expression was not available; therefore, clones were selected based on the number of copies of the FGE gene that were inserted into the genome of the cell line. No selectable markers are used as part of the process, so selection was based strictly on gene copy. Due to the possibility that overexpression of FGE could affect cell growth or protein function, clones were selected with a range of low to high gene copy numbers. Twenty-four clones were selected for further evaluation, eight with high FGE gene copy, eight with medium, and eight with low. The 24 clones were grown in culture for five weeks, and FGE level was analyzed using a flow cytometry- based assay at four different times throughout the culture. Based on FGE production and consistency of production over the extended culture, four clones were chosen for more detailed analysis (two high, one medium, and one low). These top four clones were again analyzed for FGE expression using the flow cytometry assay as well as a FGE ELISA method that was performed on cell lysates. The results indicated that one clone (#120) showed much higher expression in both assays compared with the other three clones (Figure 2, Table I). The high level of FGE expression did not significantly impact cell growth and doubling time for this clone, and it was selected as the lead candidate for future work.
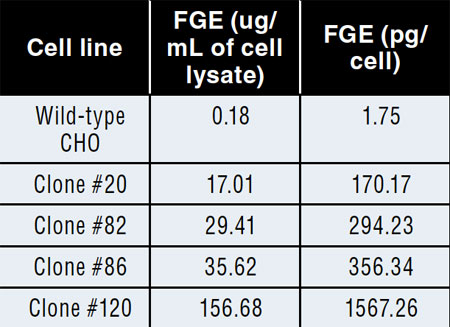
Table I: Formylglycine-generating enzyme (FGE) ELISA results from cell lysates for wild-type and FGE overexpressing cell lines.
Expression of tagged antibodies in the FGE clonal cell line
The selected FGE line was used as the base cell line to express two different antibodies. The two antibodies bind to different targets with one containing a single FGE consensus sequence and the second containing two sequences. The FGE line was engineered to produce the antibodies using the GPEx process. Limited dilution clonal selection was performed for each of the two programs and the top antibody-producing clones were selected for further analysis. These clones were evaluated in shake flasks under a nonoptimized fed-batch culture condition (containing copper [II] sulfate) measuring cell growth, antibody titer, and level of Cys to fGly conversion in the tagged sequences.
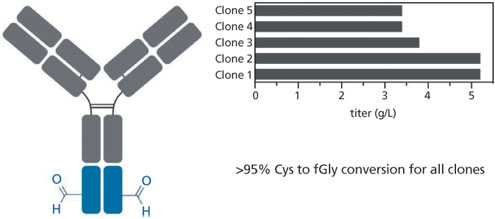
Cultures were inoculated at approximately 300,000 cells/mL and harvested when cell viabilities reached approximately 50%. For the single tagged antibody, titers for the individual clonal cell lines ranged from 3-5 g/L, with specific cell productivities as high as 75 picograms/cell/day (Figure 3). Each of these clonal lines also showed greater than 95% Cys to fGly conversion. The clones producing the double tagged antibody gave titers ranging from 1.75-2.4 g/L, and both of the sites showed high conversion rates (Figure 4).
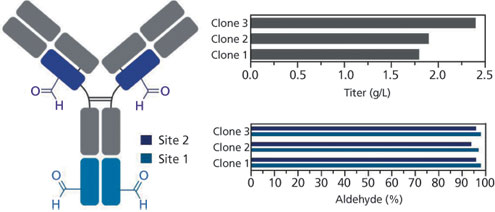 Figure 4: Non-optimized shake flask fed-batch production and conversion results of clonal cell lines producing the double-tagged antibody. Tag sites were located in CH1 domain of the heavy chain as well as at the C-terminus.
Figure 4: Non-optimized shake flask fed-batch production and conversion results of clonal cell lines producing the double-tagged antibody. Tag sites were located in CH1 domain of the heavy chain as well as at the C-terminus.
Process scale-up to bioreactors
One of the single tagged cell lines was expanded for bioreactor production to confirm that the cell lines could be scaled with similar results. Duplicate 2-L bioreactors were seeded, and the culture conditions were identical to the shake flask study, except that the reactors were harvested at approximately 80% cell viability. The duplicate reactors gave similar results to each other with antibody titers of 2.5 g/L, specific productivities of 34 picograms/cell/day, and tag conversions greater than 97% (Figure 5).

Figure 5: Duplicate 2-L bioreactor production runs under non-optimized conditions with a clonal cell producing the single tagged antibody. Specific productivities for both reactors were 34 picograms/cell/day.
To confirm that the cell line and process could be run at a typical clinical production scale, the same cell line and process was repeated at the 100L scale. The cell line and process again showed consistent results with titers reaching 2.9 g/L, a specific productivity of 43 picograms/cell/day and greater than 97% tag conversion (Figure 6).
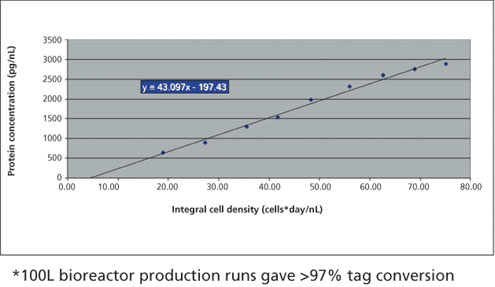
Figure 6: 100-L bioreactor production run under the non-optimized conditions with a clonal cell producing the single tagged antibody. Specific productivity for the bioreactor run was 43 picograms/cell/day.
Summary
Complex biopharmaceutical proteins in development are becoming more and more the norm with the advent of new antibody formats, unique VLP structures, and biopharmaceutical proteins that need additional processing. In the presented case study, a cell line needed to be developed that expressed three different genes (FGE, antibody heavy chain, and antibody light chain). Initially, a clonal cell line was selected that expressed FGE at high levels without impacting normal CHO cell characteristics. That base cell line was then used to produce two different tagged antibodies, both at commercially viable titers. FGE enzyme levels in those cell lines were capable of converting the antibody tag up to high specific productivities of 75 picograms/cell/day without impacting normal CHO cell behaviors in fed-batch culture conditions. Both the cell lines and culture processes were easily scalable to larger bioreactors while maintaining good antibody titers and the consistent tag conversion that is required for this unique site-specific conjugation technology, allowing for advancement into clinical development.
References
1. M.A. Frese and T. Dierks, Chembiochem 10 (3), pp. 425–427 (2009).
2. I.S. Carrico, B.L. Carlson, and C.R, Bertozzi, Nat. Chem. Biol. 3, pp. 321–322 (2007).
3. P. Wu et al., Proc. Natl. Acad. Sci. U.S.A. 106, pp. 3000–3005 (2009).
4. J.E. Hudak et al., Angew. Chem. Int. Ed. Engl. 51, pp. 4161–4165 (2012).
5. D. Rabuka et al., Nat. Protoc. 7, pp. 1052–1067 (2012).
6. P.G. Holder et al., J. Biol. Chem. 290, pp.15730–15745 (2015).
7. D. York et al., BMC Biotech. 16, pp. 23 (2016).
8. G.T. Bleck, Bioprocess J. 5, pp. 36–43 (2007).
About the Authors
Gregory Bleck is global head of R&D biologics; Dona York is director of upstream process development; and David Rabuka is global head of R&D chemical biology, all at Catalent.