This article summarizes the approaches, challenges, and future perspectives for the characterization of N-glycans in biopharmaceutical products.
By Aled Jones
 Protein glycosylation, the addition of single sugars or oligosaccharide chains (glycans) to a peptide backbone, is a common post-translational modification (PTM) that imparts various biological functions (1). More than half of all biotherapeutics are glycosylated (2), including monoclonal antibodies (mAbs), Fc-fusion proteins, clotting factors, and cytokines such as erythropoietin. Of these, mAbs represent a large proportion of biotherapeutic glycoproteins and account for approximately half of the biopharmaceutical market, a trend that is set to continue with the advent of biosimilars and biobetters (3, 4). N-Glycosylation, where glycans are attached to the nitrogen side chain of asparagine, is perhaps the most well understood form of glycosylation in biotherapeutics, particularly on mAbs, in which the N-glycan structure can affect pharmacokinetics, pharmacodynamics, and immunogenicity (5). The impact of glycosylation on protein function means that glycosylation can be a critical quality attribute (CQA), making the characterization of N-glycan structures an essential part of the biotherapeutic development process. The complex, branched structure of N-glycans present a unique analytical challenge. A variety of approaches exist, each with their own advantages and limitations. In this article, the biological effects of N-glycosylation on protein biologics are reviewed—together with the range of analytical techniques that are used to elucidate the N-glycosylation characteristics of protein biotherapeutics--and future directions for glycoanalysis are discussed.
Protein glycosylation, the addition of single sugars or oligosaccharide chains (glycans) to a peptide backbone, is a common post-translational modification (PTM) that imparts various biological functions (1). More than half of all biotherapeutics are glycosylated (2), including monoclonal antibodies (mAbs), Fc-fusion proteins, clotting factors, and cytokines such as erythropoietin. Of these, mAbs represent a large proportion of biotherapeutic glycoproteins and account for approximately half of the biopharmaceutical market, a trend that is set to continue with the advent of biosimilars and biobetters (3, 4). N-Glycosylation, where glycans are attached to the nitrogen side chain of asparagine, is perhaps the most well understood form of glycosylation in biotherapeutics, particularly on mAbs, in which the N-glycan structure can affect pharmacokinetics, pharmacodynamics, and immunogenicity (5). The impact of glycosylation on protein function means that glycosylation can be a critical quality attribute (CQA), making the characterization of N-glycan structures an essential part of the biotherapeutic development process. The complex, branched structure of N-glycans present a unique analytical challenge. A variety of approaches exist, each with their own advantages and limitations. In this article, the biological effects of N-glycosylation on protein biologics are reviewed—together with the range of analytical techniques that are used to elucidate the N-glycosylation characteristics of protein biotherapeutics--and future directions for glycoanalysis are discussed.
N-glycosylation
The flow of biological information in the DNA-RNA-protein central dogma of molecular biology (6) is complemented by a repertoire of PTMs including phosphorylation, lipidation, N-acetylation, ubiquitination, and glycosylation--the attachment of carbohydrate structures (oligosaccharides) to proteins to form glycoproteins. As of 2011, it is estimated that approximately one-fifth of the proteins on the Swiss-Prot database are glycoproteins (1). Glycosylation affects a variety of protein biological functions including folding, stability, and activity. These varied functions are enabled by the structural diversity of glycans, which stem from monosaccharide building-block composition, linkage type, and branching (7). The most prevalent forms of glycosylation are N-linked, where oligosaccharides are attached via the side-chain amino moiety (-NH2) of asparagine, and O-linked, where the sugars attach via the hydroxyl moiety (-OH) of serine or threonine. N-linked glycosylation is generally of the most frequent interest to biotherapeutic research--with a notable mention for the O-glycans present on the linker region of Enbrel (etanercept) (8). N-Glycosylation usually occurs where a protein contains the consensus sequence motif, N-X-S/T where X is not proline, although non-consensus N-linked glycosylation has been observed where the sequon is reversed (9).
What’s in a (glycan) name?
To inexperienced researchers, an initially intractable idiosyncrasy of N-glycan analysis is the number of N-glycan nomenclatures in current use. This can complicate interactions between scientists, as common N-glycan structures may have alternate names and diagrammatic representations. For example, arguably the most common N-glycan structure present on mAbs made in Chinese hamster ovary (CHO) cells is G0F: asialo-, agalacto-, biantennary complex N-glycan, core-substituted with fucose. This glycan is also known as G0, FA2, and F(6)A2. The latter name uses “Oxford” nomenclature (10), which has an associated system for generating structural diagrams of glycans. The other main system for “cartoons” of glycan structures is from the Consortium for Functional Glycomics (CFG) (11). Each has their advantages: Oxford structures convey linkage type (α or β) and position without the use of text; CFG structures are more spatially compact. Either Oxford or CFG structures can be drawn with GlycoWorkbench software (EUROCarbDB) (12), and CFG structures are used here.
N-glycans of antibody and antibody-derived biotherapeutics
The influence of N-glycans on the efficacy, stability, half-life, and safety of antibody and antibody-derived biotherapeutics are well-reviewed (5, 13, 14) and are summarized in Figure 1. Immunoglobulin G (IgG) molecules consist of two heavy and two light chains, and the heavy chains contain an N-linked glycosylation site at asparagine 297 within the fragment crystallizable (Fc) region. In addition, some IgGs such as Erbitux (cetuximab) are also N-glycosylated within the fragment antigen-binding (Fab) region. Figure 1A represents some of the therapeutic effects of biantennary N-glycans of the Fc region of IgGs. Interaction of the Fc region of IgG with Fc-γ receptors (FcγR) influences immune effector functions including antibody-dependent cell-mediated cytotoxicity (ADCC) by binding to Fcγ RIIIa, a mechanism of action of some therapeutic IgGs. The absence of fucose linked to the innermost GlcNAc of Fc N-glycans (“core” fucose) enhances binding to Fcγ RIIIa and ADCC activity (15), contrasting with the lower activity of mostly fucosylated mAbs produced in CHO cells. Consequently, glycoengineereing approaches have been developed to generate afucosyl antibodies with enhanced ADCC activity, including α-1,6-fucosyltransferase (FUT8) knockout CHO cell lines (16) and inhibitors of fucosylation (17). Galactosylation can also enhance Fcγ RIIIa binding, potentially through conformational changes (18), and is additive to the ADCC effect of afucosylation. Terminal galactose also increases binding to complement 1q (C1q), leading to enhanced complement-dependent cytotoxicity (CDC) (14). Terminal sialic acid is anti-inflammatory, particularly when linked α(2,6) to galactose as opposed to the α(2,3) linkage produced by CHO cells (19). Presence of sialic acid residues may also reduce ADCC activity, and reduces clearance by shielding galactose from asialoglycoprotein receptors.
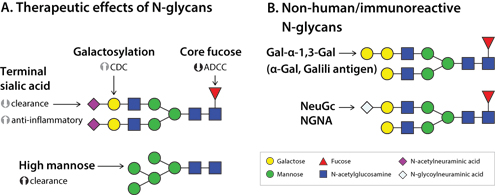 Figure 1: Structure-function relationship of biotherapeutic N-glycans. A) Therapeutic effect of N-glycan structure on Fc and Fc-derived molecules. B) Immunogenicity of N-glycan structures. ADCC=antibody-dependent cell-mediated cytotoxicity; CDC=complement-dependent cytotoxicity; NGNA=N-Glycolylneuraminic acid.
Figure 1: Structure-function relationship of biotherapeutic N-glycans. A) Therapeutic effect of N-glycan structure on Fc and Fc-derived molecules. B) Immunogenicity of N-glycan structures. ADCC=antibody-dependent cell-mediated cytotoxicity; CDC=complement-dependent cytotoxicity; NGNA=N-Glycolylneuraminic acid.
High mannose glycans represent an immature glycan structure, and may be the result of suboptimal CHO cell-culture conditions (20). The presence of high-mannose glycans results in increased clearance mediated by mannose receptors (14), and as such, when ADCC constitutes the mechanism of action, the presence of high-mannose-type N-glycans is a CQA.
As shown in Figure 1B, there are non-human or immunoreactive N-glycan structures that may be present on biotherapeutics produced in vitro. The α-gal epitope, or Galili antigen, is formed when galactose is α1,3-linked to galactose, and is a structure not produced by humans (21). α-gal has been associated with IgE-mediated hypersensitivity reactions to Erbitux (cetuximab) produced in the mouse myeloma line SP2/0, where there are α-gal N-glycans on the Fab region (22). CHO cells are also capable of producing the α-gal epitope (23), although interestingly, IgE binding is not observed when α-gal is present on Fc N-glycans of Remicade (infliximab) and Orencia (abatacept) (24). N-Glycolyneuraminic acid (NGNA or Neu5Gc) is a sialic acid not synthesized by humans and is, therefore, potentially immunogenic; it is found in biotherapeutic proteins probably due to the use of non-human mammalian cell lines and the use of animal-derived components of culture media (25).
Approaches to N-glycan analysis
Figure 2 illustrates the levels of analytical approaches to N-glycan analysis, from intact protein through to glycopeptides, released glycans, and down to individual monosaccharide components (2, 26). The intact mass of the different glycoforms of a glycoprotein can be characterized by high-resolution accurate mass-mass spectrometry (HRAM-MS), coupled to reverse-phase liquid chromatography (RP-LC). This “top-down” approach requires minimal sample preparation, although reduction can be used to analyze heavy and light IgG chains separately, or can be fragmented with specific proteases, such as IdeS. LC-ESI-MS has also been used to analyze the glycosylation of erythropoietin (EPO), including darbepoetin alfa, an EPO analog with two additional N-linked glycan sites and a longer serum half-life (27). Capillary electrophoresis (CE)-electrospray ionization (ESI)-mass spectrometry (MS) (CE-ESI-MS) may also be used to profile intact glycoform masses (28).
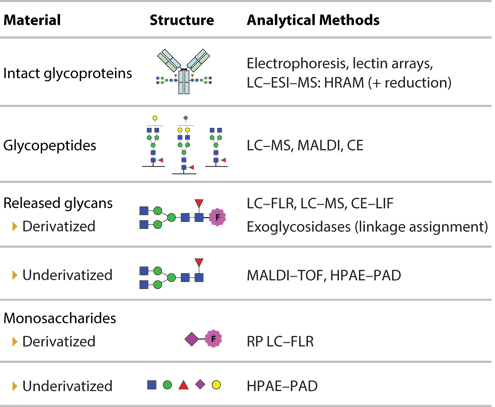
Figure 2: Analytical approaches to N-glycan analysis. LC=liquid chromatography; ESI-MS=electrospray ionization mass-spectrometry; HRAM=high resolution, accurate mass; CE=capillary electrophoresis; FLR=fluorescence; LIF=laserinduced fluorescence; RP=reverse-phase; HPAE-PAD=high performance anionexchange chromatography with pulsed amperometric detection.
To determine glycosylation site occupancy, glycoproteins may be broken down into glycopeptides with tryspin or other proteases, and analyzed by LC-MS or CE-MS. However, while glycopeptide analysis provides compositional information on glycans, it does not always provide structural information on linkage and branching (29).
N-glycan release
Analysis of N-glycans is assisted by the existence of several enzymes that can release N-glycans. Of these, the most well-known is peptide-N-glycosidase F (PNGase F), an amidase that releases intact complex bianennary, high-mannose and hybrid-type N-glycans by cleaving between the innermost GlcNAc of the N-glycan and asparagine, unless the GlcNAc is α1,3-fucosylated, in which case PNGase A must be used. PNGase F digestion time has recently been reduced from hours to minutes in recent protocols (30, 31), dramatically shortening the N-glycan sample preparation workflow. PNGase A releases N-glycans that are α1,3-fucosylated.
Endoglycosidases such as EndoS2 may also be used, to take advantage of the fact that this enzyme cleaves between the two GlcNAcs in the N-glycan core, leaving a single afucosyl or fucosyl GlcNAc attached to the protein. A published workflow described the use of endoglycosidase EndoS2 in combination with IdeS and glycopeptide analysis for quantitative determination of afucosylation (32).
Unfortunately, the O-glycan field does not benefit from similar enzymes that can release intact O-glycans. O-Glycanase (endo-α-N-acetylgalactosaminidase) can only release Gal-GalNAc from serine or threonine; it cannot release extended O-glycan structures. O-glycan release can be performed with hydrazinolysis or beta elimination, and a recent study has made use of household bleach for this purpose (33). Chemical release can also be used for N-glycan analysis, but is less frequently employed due to the existence of PNGase F.
N-glycan labeling
Released N-glycans are frequently labeled with a fluorophore to enhance glycan separation and detection (34). Commonly used labels include 2-aminobenzamide (2-AB) and 2-aminobenzoic acid (2-AA, also known as anthranilic acid) for LC separations, 8-Aminonaphthalene-1,3,6-trisulfonic acid (ANTS) for fluorophore-assisted carbohydrate electrophoresis (FACE), and 1-aminopyrene-3,6,8-trisulfonic acid (APTS) for capillary electrophoresis (CE). However, these dyes use reductive amination chemistry, which requires heated incubation. Fluorophores including InstantPC (ProZyme) (30) and RapiFluor-MS (Waters) (31) have been developed that can label the glycosylamine produced immediately after PNGase F digestion--thus, speeding up the workflow--and these dyes have enhanced fluorescence and MS properties compared with 2-AA and 2-AB.
Separation of N-glycans by liquid chromatography
Hydrophilic interaction liquid chromatography (HILIC) is a widely accepted technique for the separation of labeled N-glycans (35), and the advent of ultra-high performance liquid chromatography (UHPLC) has improved resolution and speed. HILIC is often coupled with detection by fluorescence, resulting in a chromatographic profile that can be used to determine the relative abundance of N-glycan species present in the sample. Figure 3 shows an example of released N-glycans from Enbrel labeled with InstantPC and separated by HILIC with fluorescence detection. HILIC can also be coupled to MS (36) to aid in structure identification. Depending on the separation, linkage isomers may also be identified by the profile: N-glycans with α(2,3)-sialylation have shorter HILIC retention times than isomeric N-glycans with α(2,6) sialic acid linkages (19). Labeled N-glycans may also be digested with exoglysocidases prior to separation to facilitate peak assignment. For example, sialic acid linkage position may also be determined by exoglycosidase digests with sialidases (also known as neuraminidases). Commercially available sialidase from Streptococcus pneumoniae releases non-reducing terminal α(2,3)-linked sialic acid, and sialidase from Arthrobacter ureafaciens releases α(2-3,6,8,9)-linked sialic acid. Digestion of glycans with alkaline phosphatase prior to separation has been shown to identify glycans bearing mannose-6-phosphate (M6P), an important glycan epitope for lysosomal targeting of enzyme replacement therapies (37).
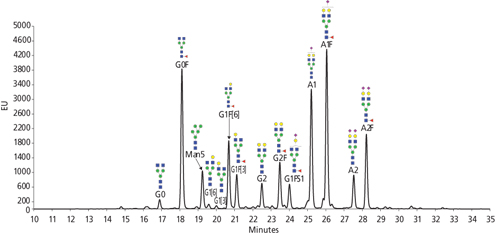
Figure 3: Released N-glycans from Enbrel labeled with InstantPC and separated by hydrophilic interaction liquid chromatography (HILIC) with fluorescence detection. Data courtesy John Yan, ProZyme.
Capillary electrophoresis of N-glycans
N-Glycans labeled with a charged fluorophore may be separated by CE (40) and detected by laser-induced fluorescence (LIF), LED-induced fluorescence (LEDIF), or MS (41). The selectivity of CE is useful for analysis of N-glycans containing the α-gal epitope, as they migrate more slowly than other neutral glycans and sialylated glycans (42). Recent developments in CE have allowed for rapid analysis, enabled by short run time on the Gly-Q (ProZyme) (43) or multiplexed analyses on the GlycanAssure (Thermo Fisher Scientific) (44) and GlyXera (45) instruments. These new platforms are coupled with software to assist in peak integration, peak calling, and determination of relative abundance, allowing many samples to be processed in a single day. Ease and speed of analyses make it more attractive to examine N-glycan composition in the earlier stages of biotherapeutic process development such as during clone selection and screening, where samples may be more numerous.
Monosaccharide analysis
Finally, it is also possible to obtain compositional information of glycans by breaking them down into their monosaccharide constituents with acid hydrolysis and separating the monosaccharides with HPAE-PAD (46), yielding information on the relative amounts of each monosaccharide. Other techniques for monosaccharide analysis include conjugation of sialic acids to 1,2-diamino-4,5-methylene-dioxybenzene (DMB), gas chromatography with MS (GC-MS), and HPLC (47).
Future directions in N-glycan analysis
As glycosylation is a frequently monitored CQA of biotherapeutics, there will likely be increasing regulatory pressure to control processes for better safety, efficacy, and product quality, compounded by the advent of patent expirations and the emergence of biosimilars and biobetters. Advances in glycoanalysis and glycoengineering will be key to informing research, development, and manufacturing decisions. With glycan research progressing at an accelerated pace over the past two decades, N-glycan analysis is set for continued advancement in three areas: sample preparation; glycan separation and detection; and data analysis. For sample preparation, increased speed and automation are likely to continue the trend of recent developments in shortening preparation time from days to hours, and enzymatic fragmentation of biotherapeutic proteins into constituent domains will enhance the level of data obtained from a single set of samples. The use of reference-standard glycoprotein material such as The National Institute of Standards and Technology’s NISTmAb (48) is likely to help unify approaches across laboratories analyzing glycosylated biotherapeutics. Similarly, glycan separations by LC and CE are set to speed up while maintaining or improving resolution. Given the advances in sample preparation and glycan separation, the need to easily analyze voluminous datasets will bring analytical tools into focus, involving software engineers, computer scientists, chemists, and biologists across industry and academia. Combined, these factors could power glycomic analyses at a similar rate as the rate that was seen with the advancement of genomic and proteomic technologies during the past 20 years.
References
1. G.A. Khoury, R.C. Baliban, and C.A. Floudas, Nat. Sci. Rep. 1 (90), doi: 10.1038/srep00090 (2011).
2. A. Planinc et al., Anal. Chem. Acta 921, 13-27 (2016).
3. G. Walsh, Nat. Biotechnol. 32 (10), 992-1000 (2014).
4. D.M. Ecker, S.D. Jones, and H.L. Levine, mAbs 7 (1), 9-14 (2015).
5. L. Liu, J. Pharm. Sci. 104 (6), 1866-1884 (2015).
6. F. Crick, Nature 277, 561-562 (1970).
7. C.R. Bertozzi and D. Rabuka, “Structural Basis of Glycan Diversity,” in Essentials of Glycobiology, 2nd edition, A. Varki et al., Eds. (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 2nd ed., 2009), 23-36.
8. S. Houel et al., Anal. Chem. 86, 576-584 (2014).
9. J.F. Valliere-Douglass et al., J. Biol. Chem. 285 (21), 16012-16022 (2010).
10. D.J. Harvey et al., Proteomics 9 (15), 3796-801 (2009)
11. A. Varki et al., Proteomics 9 (24), 5398-5399 (2009).
12. A. Ceroni et al., J. Proteome Res. 7 (4), 1650-1659 (2008).
13. F. Higel, et al., Eur. J. Pharm. Biopharm. 100, 94-100 (2016).
14. D. Reusch and M.L. Tejada, Glycobiology 25 (12), 1325-1334 (2015).
15. T. Shinakawa et al., J. Biol. Chem. 278, 3466-3473 (2003).
16. A. Beck and J.M. Reichert, mAbs 4 (4), 419-425 (2012).
17. N.M. Okeley et al., Proc. Natl. Acad. Sci. U.S.A. 110 (14), 5404-5409 (2013).
18. D. Houde et al., Mol. Cell Prot. 9 (8), 1716-1728 (2010).
19. C. Raymond et al., mAbs 7 (3), 571-583 (2015).
20. Y. Fan et al., Biotechnol. Bioeng. 112 (3), 521-535 (2015).
21. B.A. Macher and U. Galili, Biochim. Biophys. Acta 1780 (2), 75-88 (2008).
22. C.H. Chung et al., N. Engl. J. Med. 358 (11), 1109-1117 (2008).
23. C.J. Bosques et al., Nat. Biotechnol. 28 (11), 1153-1156 (2010).
24. J.L. Van Bueren et al., Nat. Biotechnol. 29, 574-576 (2011).
25. D. Ghaderi et al., Biotechnol. Genet. Eng. Rev. 28, 147-175 (2012).
26. L. Zhang, S. Luo, and B. Zhang, mAbs 8 (2), 205-215 (2016).
27. A. Harazono et al., J. Pharm. Biomed. Anal. 83, 65-74 (2013).
28. R. Haselberg, G.J. de Jong, and G.W. Somsen, Anal. Chem. 85, 2289-2296 (2013).
29. H. Desaire, Mol. Cell Prot. 12 (4), 893-901 (2013).
30. M. Lauber et al., Anal. Chem. 87 (10), 5401-5409 (2015).
31. A. Jones, “N-Glycan Analysis using InstantDye Workflows for the Screening & Characterization of Biotherapuetics,” presentation at the WCBP Conference (Washington, DC, 2017).
32. R. Upton et al., Anal. Chem. 88 (20), 10259-10265 (2016).
33. X. Song et al., Nat. Methods 13, 528-534 (2016).
34. L.R. Ruhaak et al., Anal. Bioanal. Chem. 397 (8), 3457-3481 (2010).
35. D. Reusch et al., mAbs 7 (1), 167-179 (Jan.-Feb. 2015).
36. D. Reusch et al., mAbs 7 (4), 732-742 (Jul.-Aug. 2015).
37. J. Bones et al., Anal. Chem. 83 (13), 5344-5352 (2011).
38. G.C.M. Vreeker and M. Wuhrer, Anal. Bioanal. Chem. 409 (2), 359-378 (2017).
39. Z. Szabo et al., Anal. Bioanal. Chem. 409 (12), 3089-3310 (2017).
40. Á. Szekrényes et al., mAbs 8 (1), 56-64 (2016).
41. R.G. Jayo et al., Anal. Chem. 86 (13), 6479-6486 (2014).
42. Z. Szabo et al., Mol. Pharm. 9 (6), 1612-1619 (2012).
43. ProZyme, Inc., “ProZyme, Inc. Launch Gly-Q System for Integrated N-Glycan Analysis,” Press Release (Hayward, CA, November 2016).
44. A. Eckhardt, “Rapid and Robust N-Glycan Profiling Workflow for Therapeutic Proteins with UHPLC and Multicapillary CE3500xL,” WCBP Conference (Washington, DC, 2017).
45. R. Hening et al., Biochim. Biophys. Acta 1860 (8), 1728-1738 (2016).
46. J.S. Rohrer, L. Basumallick, and D. Hurum, Biochemistry (Moscow, Russ. Fed.) 78 (7), 697-709 (2013).
47. B. Mulloy, G.W. Hart, and P. Stanley, “Structural Analysis of Glycans,” in Essentials of Glycobiology, 2nd edition, A. Varki et al., Eds. (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 2nd ed., 2009), Chapter 47.
48. J.M. Prien et al., “Orthogonal Technologies for NISTmAb N-Glycan Structure Elucidation and Quantitation,” in State-of-the-Art and Emerging Technologies for Therapeutic Monoclonal Antibody Characterization, Volume 2. Biopharmaceutical Characterization: The NISTmAb Case Study, J.E. Schiel et al., Eds. (American Chemical Society, 2016), 185-235.