The Evolving Role of Starting Materials in Cell and Gene Therapy
 Accelerated approval pathways and growing demand for cell and gene therapies are putting pressure on providers of cellular starting materials, and they must ensure a steady supply.
Accelerated approval pathways and growing demand for cell and gene therapies are putting pressure on providers of cellular starting materials, and they must ensure a steady supply.
The number of product approvals for cell and gene therapies is rising steadily. Earlier this year, FDA issued a statement revealing that there are already more than 800 active cell-based or directly administered gene therapy investigational new drug applications (INDs) currently on file, and that by 2020, they expect to be receiving more than 200 INDs per year (1). Based on this forecast, the industry expects cell and gene therapy products to surge in the market and remain a prominent part of the pharmaceutical industry for years to come (2).
FDA has developed four distinct pathways to speed the progress of promising new therapies through clinical trials, and if results merit, on to approval. Advanced therapies may be eligible for one or more of these designated pathways if they treat a serious condition, address unmet medical needs, or obtain early clinical results that show significantly improved outcomes compared to current therapies. In 2018, FDA’s Center for Drug Evaluation and Research (CDER) issued a record-breaking 59 new drug approvals (3). Of these new approvals, 32% were authorized as first-in-class, and 58% were approved to treat rare or “orphan” diseases.
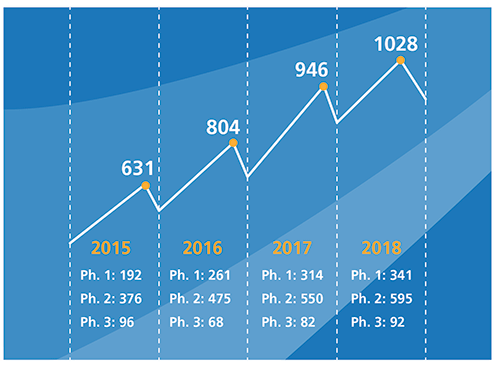 Figure I. Phase I to III clinical trials over a four-year period.There are three cell therapy products that have been approved thus far: Provenge (sipuleucel-T) from Dendreon Pharmaceuticals, Kymriah (tisagenlecleucel) from Novartis, and Yescarta (axicabtagene ciloleucel) from Kite Pharma, a Gilead Sciences company. These are likely to be the tip of the iceberg. Because many new cell and gene therapies are targeting unmet medical needs, orphan diseases, or both, they are much more likely to request (and receive) accelerated approval. The Alliance for Regenerative Medicine reports that as of the end of 2018, 263 cellular therapies and 362 gene-modified cellular therapies were being evaluated in clinical trials (see Figure 1) (4). The majority of these clinical trials are for cancer indications, with chimeric antigen receptor (CAR)-T cell therapies alone accounting for 181 Phase I–III clinical trials (5).
Figure I. Phase I to III clinical trials over a four-year period.There are three cell therapy products that have been approved thus far: Provenge (sipuleucel-T) from Dendreon Pharmaceuticals, Kymriah (tisagenlecleucel) from Novartis, and Yescarta (axicabtagene ciloleucel) from Kite Pharma, a Gilead Sciences company. These are likely to be the tip of the iceberg. Because many new cell and gene therapies are targeting unmet medical needs, orphan diseases, or both, they are much more likely to request (and receive) accelerated approval. The Alliance for Regenerative Medicine reports that as of the end of 2018, 263 cellular therapies and 362 gene-modified cellular therapies were being evaluated in clinical trials (see Figure 1) (4). The majority of these clinical trials are for cancer indications, with chimeric antigen receptor (CAR)-T cell therapies alone accounting for 181 Phase I–III clinical trials (5).
While a robust pipeline is certainly a strong indicator of future therapeutic success, the rapid increase in the number of cell-based therapies headed toward commercialization is placing growing demands on cell-therapy starting material suppliers. Unlike other biological drug products such as therapeutic proteins, monoclonal antibodies, or vaccines, drugs based on living cells are by nature uniquely vulnerable to supply chain complications and setbacks. Apheresis center capacity, donor network access, sample handling expertise, and quality control systems all contribute to the ability to achieve reproducible starting material quality and efficacy.
| Cell therapy starting material—where does it feed into critical process points? |
|---|
| Donor recruitment: Necessary at each stage of clinical testing. Must have a strong donor network and viable back-up candidates. |
| Critical quality attribute identification/assay development: Identify and develop assays for critical quality attributes, potency assays—overestimate testing needs! |
| Methods/equipment qualification: Methodology and equipment/reagents tested for quality control and to minimize contamination risks. |
| Process qualification: All purification/isolation and processing steps should be optimized for target cell type. |
| Scale-up/scale-out:Stability studies and cryopreservation/biopreservation methods optimization, logistics worked out. |
| Manufacturing change validation/product comparability studies:Necessary to re-validate process with starting material that is essentially indistinguishable from original material in terms of quality, potency. |
Starting material variability
The raw materials for allogeneic cell therapies cannot be manufactured on demand, but instead, rely on a network of volunteer donors. This creates variability in cell therapy starting material from the outset. Small donor networks are vulnerable to issues, such as unexpected drops in the number of donations, uneven reliability, and inadequate diversity to meet the specific needs of their clients. Sourcing cells from a large, diverse donor network avoids many of these complications, but cell-therapy manufacturing and supply companies must still deal with the inherent variability of living cells and tissues.
Many different factors can introduce variability into the total yield of white blood cells (WBC) and other therapeutic cells during apheresis collection. Disease, for example, impacts cellular functional capabilities and overall cell health. Cells collected from patients with illness display significant physiological differences compared to those collected from healthy donors. Cell subset composition, cell phenotype expression, and the presence of inflammatory or stress-related factors can all impact target cell health, function, yield, and downstream therapeutic potency.
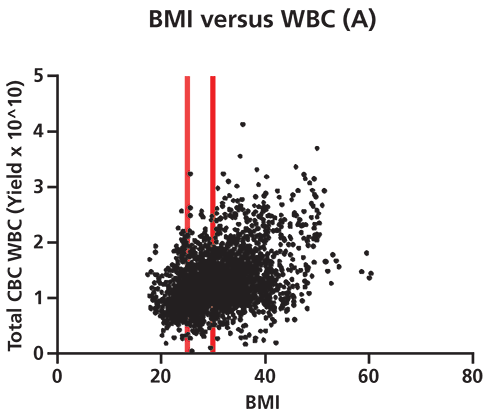
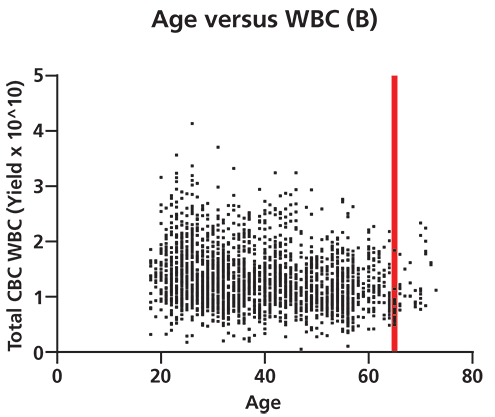 Figure 2. Differences in white blood cell (WBC) yield correlate to donor biometrics such as age and body mass index (BMI). A shows that there is a significant positive correlation (95% confidence by Spearman correlation test) between donor BMI and total WBC yield per apheresis collection unit. This correlation grows more pronounced as BMI increases from normal (cutoff at first red line) to overweight (cutoff at 2nd red line) to obese in terms of medically defined weight categories. p=<0.0001, two-tailed t-test. B shows that there is a significant negative correlation between donor age and total WBC yield per apheresis collection. This correlation grows more pronounced beyond age 65 (denoted by red line, 95% confidence level by Spearman correlation). p=<0.0001, two-tailed t-test (n=2490 cell collections). CBC is complete blood count. The collection of healthy donor cells is also subject to considerable variability, and a well-established donor center will be aware of the many factors that impact WBC counts (6). Data generated illustrates the variability present in apheresis collections from normal healthy donors (Figure 2). For example, WBC yields vary significantly with donor body mass index (BMI) and advanced age. Factors such as age, gender, BMI, ethnicity, current medications, and medical history have all been documented to result in differences in WBC profiles (7, 8). Stressful personal issues and lifestyle habits such as smoking, drinking to excess, poor sleep habits, and poor nutrition all impact WBC counts. Good donor communication can give cell collection center staff valuable knowledge about which factors known to affect target cell collection may be present, as well as the opportunity to modify cell collection protocols appropriately.
Figure 2. Differences in white blood cell (WBC) yield correlate to donor biometrics such as age and body mass index (BMI). A shows that there is a significant positive correlation (95% confidence by Spearman correlation test) between donor BMI and total WBC yield per apheresis collection unit. This correlation grows more pronounced as BMI increases from normal (cutoff at first red line) to overweight (cutoff at 2nd red line) to obese in terms of medically defined weight categories. p=<0.0001, two-tailed t-test. B shows that there is a significant negative correlation between donor age and total WBC yield per apheresis collection. This correlation grows more pronounced beyond age 65 (denoted by red line, 95% confidence level by Spearman correlation). p=<0.0001, two-tailed t-test (n=2490 cell collections). CBC is complete blood count. The collection of healthy donor cells is also subject to considerable variability, and a well-established donor center will be aware of the many factors that impact WBC counts (6). Data generated illustrates the variability present in apheresis collections from normal healthy donors (Figure 2). For example, WBC yields vary significantly with donor body mass index (BMI) and advanced age. Factors such as age, gender, BMI, ethnicity, current medications, and medical history have all been documented to result in differences in WBC profiles (7, 8). Stressful personal issues and lifestyle habits such as smoking, drinking to excess, poor sleep habits, and poor nutrition all impact WBC counts. Good donor communication can give cell collection center staff valuable knowledge about which factors known to affect target cell collection may be present, as well as the opportunity to modify cell collection protocols appropriately.
Apheresis and healthcare experts should understand donor variability issues and be trained to optimally match the unique physiological characteristics of each donor with the project needs of the client. They also need the training and expertise to operate and maintain apheresis instrumentation and to decide which collection methods best suit the donor and client. Differences in staff training and apheresis equipment and supplies can also introduce variability. Standardizing equipment and training between the manufacturers’ various collection sites can alleviate starting material variability and downstream processing risk.
Safeguarding quality
Along with donor-related variability, collection methods and post-collection sample handling also contribute to starting material variability. It is all but inevitable that a small percentage of cells will be lost to purification or target cell modification steps, which is why it’s best to develop a process that is as integrated as possible. Automation and closed-loop systems can cut down on process related cell loss.
Flawed sample handling during cell collection and transfer to the manufacturing facility, however, carries a greater risk than minor process losses. Contamination risk, whether from outside sources or intrinsic contamination from other cell types, is an ongoing concern for cell therapy developers because it can result in serious medical and financial consequences. Risks of this type can be avoided through stringent oversight of good manufacturing practices (GMP)-compliant cleanrooms and isolator hoods as well as through the use of closed culturing systems and automated, inline quality attribute monitoring.
Contamination isn’t the only risk to starting material quality. Cells are capable of responding to their environment, and exposing cells to physiological stress factors can lead to apoptosis and loss of target cell function and potency.
For years, the mesenchymal stem cell (MSC) therapy field struggled with intermittent loss-of-function issues, despite promising clinical trial results for treating a host of different diseases. Several research groups reported that the cells were vulnerable to loss of viability and function following cryopreservation and suggested that it might only be possible to use MSCs that were freshly isolated (9). Because this would present obvious logistical problems for a commercialized therapy, some intense investigation followed. It was eventually determined that MSCs are particularly susceptible to heat stress during cell thawing. Post-thaw recovery of MSCs in culture for a 24-hour period allows these cells to fully regain their functional potency (10).
Cell therapy source material losses resulting from sub-optimal cell preservation and storage techniques can result in bottlenecks that slow the entire manufacturing process. Biopreservation and cryopreservation cocktails, as well as cell freezing and thawing methods, can have an enormous impact on product quality and efficacy. Similarly, shipping and storage logistics can introduce risk to these vulnerable products in the form of unforeseen delays or inadequate temperature regulation, which can affect product stability, again leading to loss of viability and functionality. Because cryopreservation of therapeutic cells is central to their successful commercialization, cryopreservation and shipping logistics need to be optimized as early as possible during process development.
Planning for success
Cell sourcing and variability are two of the most important challenges facing the fledgling cell therapy industry. Inconsistent access or inconsistent starting material quality can leave manufacturers shorthanded, increasing the risk of batch failure or missed production deadlines. With increased pressure on hospitals and other apheresis centers to procure greater quantities of high-quality target cells, these centers are scrambling to increase yields without sacrificing quality.
While ideally all donors would be able to donate higher blood volumes rich in the target cell population, realistically, donors with these characteristics are not always readily available. Manufacturers need to incorporate collection methods that help counter issues such as low volume yields or the presence of contaminating cell types.
Cell collection efficiency refers to the ratio of the target cell population to contaminating cell types present in the apheresis product as well as the ratio of total yield of target cells collected to those calculated to be present in the donor’s circulation. Because contaminating cell types can slow or complicate downstream isolation protocols, yield and purity should be balanced with the goal of consistently delivering maximum therapeutic efficacy to the patient.
The best way to protect cellular starting material quality is to have strong quality management systems in place from start to finish (11). Real-time monitoring and quality control checkpoints using validated, physiologically relevant functional potency assays are a necessary part of the process. Cell collection and processing centers need to be tightly coordinated to prevent loss of efficacy due to raw material transfer logistics.
As the industry evolves, steps are being taken to improve access to donor materials and ensure their consistent quality. Cell collection centers and their pharmaceutical manufacturing partners need to integrate and coordinate their efforts early in the development process. Establishing and maintaining large, diverse donor networks involves proactive outreach to potential donors by experienced, highly trained staff. Training should include expertise in donor selection and an understanding of the impact of demographics and lifestyle characteristics on cell yield and purity. It should also cover collection methods, instrumentation, data collection, and biological sample handling and storage.
Discussion
Cell and gene therapy companies are still newcomers to the medical field. As new products start emerging onto the scale-up and marketing stage, manufacturing and supply companies are actively building the expertise and infrastructure necessary for commercial success.
Issues such as starting material sourcing and quality validation will require the integration of a number of different strategies. One of the most important strategies manufacturers need to implement is investing significant resources early on for intensive product testing and characterization, including characterization of apheresis materials that are the critical foundation for the product.
Discerning which quality attributes are the most critical and developing quality control assays that accurately assess these attributes throughout process development and manufacturing are crucial to commercial success. In some ways this is an ongoing process; scale-up may necessitate manufacturing changes that impact which quality attributes are the most critical to track, or whether further critical quality attribute (CQA) assays need to be appended to the quality control systems. This is where exhaustive product characterization pays off. Understanding the optimal operating limits of critical reagents and methods can speed process adaptation and product re-validation.
Cell-therapy starting material sourcing is a tremendous challenge, but one that can be mitigated through a variety of integrated efforts. Hospitals and apheresis clinics are set up to handle patients; they are generally not prepared to accommodate increased demand for cell therapy starting materials. While one cannot always avoid a hospital setting for the collection of patient donor materials, perhaps a new paradigm is necessary to transition away from hospitals as the main resource for healthy donor materials, and toward apheresis suppliers whose business model is specifically geared toward supporting cell and gene therapy development.
Some larger-scale apheresis material collection centers are already incorporating the preparation of GMP-compliant materials on site, integrating cell collection with GMP-compliant preliminary processing (12). This provides the benefit of regulatory compliance and assured quality from the outset, while also avoiding the need to transport collected apheresis products to an off-site facility for preliminary characterization, processing, and target cell isolation procedures.
The development and standardization of suitable sample handling protocols based on tissue and cell type is another critical support issue for both cell-therapy starting materials and final product. Automation, process integration, closed-system design, and compliance with current GMP can all be used to safeguard cell-therapy product quality.
Regulatory guidelines issued by FDA and other regulatory agencies are still evolving to suitably accommodate advanced medicinal products such as cellular therapies. Feedback from newly approved products has led to an increased emphasis on commercial preparedness on the part of cell therapy companies seeking expedited approval in order to assure patients are protected from supply chain shortages (13). As the cell and gene therapy industry matures, manufacturing and supply companies are putting in place the physical infrastructure and expertise needed to minimize variability and expand starting-material sourcing capabilities. Standardizing quality control systems and nomenclature, strengthening raw material and reagent characterization, and optimizing sample handling methods will significantly reduce risk for the industry. Close cooperation between research and development labs, regulatory agencies, apheresis supply centers, and manufacturing facilities will drive successful commercialization of these much-needed therapies.
References
1. FDA, “Statement from FDA Commissioner Scott Gottlieb, MD, and Peter Marks, MD, PhD, Director of the Center for Biologics Evaluation and Research on New Policies to Advance Development of Safe and Effective Cell and Gene Therapies,” Press Release, Jan. 15, 2019.
2. A. Philippidis, Genetic Engineering and Biotechnology News 39 (7) (2019).
3. FDA, “2018 New Drug Therapy Approvals,” www.fda.gov/media/120357/download [3], 2019.
4. L. Scull, “Cell & Gene Therapy Sector Overview [4],” Presentation for the Alliance for Regenerative Medicine, Feb. 27, 2019.
5. ARM, “Regenerative Medicine & Oncology [5],” June 2019.
6. B. Taylor and D. Clarke, “Why is Healthy Donor Tissue So Important for Cell and Gene Therapies?” Hemacare.com, June 27, 2019.
7. E. Nah., et al., Ann Lab Med. 38 (6) 503–511 (2018).
8. L. Jamshidi and A. Seif, J Res Med Sci. 19 (2) e4955 (2017).
9. R. Chinnadurai, et al., Stem Cells 34 (9) 2429–2442 (2016).
10. B. Antebi, et al. Journal of Translational Medicine 17, 297 (2019).
11. D. Clarke and B. Taylor, European Biopharmaceutical Review, pp. 24–28 (2019).
12. HemaCare, “HemaCare Details Expanded GMP Capabilities at CAR-TCR Summit in Boston, MA.,” Press release, Sept. 4, 2018.
13. C. Bennett, “Cell Therapy Manufacturing: The Supply Chain Challenge,” GenEngNews.com, Nov.16, 2018.
About the authors
Brad Taylor*, PhD, [email protected] [6], is global marketing director, and Dominic Clarke, PhD, is global head of cell therapy, both at Hemacare Corporation, 8500 Balboa Boulevard, Suite 130, Northridge, CA 91325.
*To whom correspondence should be addressed.