The authors review the status of expression of antibodies in microbial hosts and present the recent advances in the production of aglycosylated antibodies in bacteria.
By Anurag S. Rathore, Jyoti Batra
 The biopharmaceutical industry is growing exponentially, driven by an ever-increasing demand for monoclonal antibodies (mAbs) and related products that are capable of treating complex, life-threatening diseases such as cancer, as well as other infectious and autoimmune diseases. With more than 200 recombinant proteins on the market today, the biopharmaceutical industry is expected to reach approximately $180 billion by 2016 (1). Therapeutic mAbs and their derivatives—such as antibody fragments (Fab), single-chain Fv (scFv), and diabodies—represent the fastest-growing class of approved therapeutic proteins because of their high specificity, increased serum half-life, and low toxicity (2). These products have been used in biosensors, protein purification, and bioimaging (3).
The biopharmaceutical industry is growing exponentially, driven by an ever-increasing demand for monoclonal antibodies (mAbs) and related products that are capable of treating complex, life-threatening diseases such as cancer, as well as other infectious and autoimmune diseases. With more than 200 recombinant proteins on the market today, the biopharmaceutical industry is expected to reach approximately $180 billion by 2016 (1). Therapeutic mAbs and their derivatives—such as antibody fragments (Fab), single-chain Fv (scFv), and diabodies—represent the fastest-growing class of approved therapeutic proteins because of their high specificity, increased serum half-life, and low toxicity (2). These products have been used in biosensors, protein purification, and bioimaging (3).
Since the advent of an era of antibody-based therapeutics, the major focus of the industry has been on the generation of antibodies with glycosylation patterns similar to the naturally occurring immunoglobulin Gs (IgGs). Mammalian cells have been primarily applied as the hosts of choice for mAb expression. However, clinical success of mAbs has tremendously increased their demand, and this in turn has fueled interest in other cost-effective alternative production systems such as microbial hosts. Further, mAb production in microbial hosts such as bacterial cells completely abrogates the nuisance of glycosylation control and speeds up production timelines, further simplifying bioprocessing. The groundbreaking discovery of hybridoma cell generation technology leading to mAb production is one of the key developments in the past decade (4). Generation of antibodies in microbial hosts was initially met with skepticism due to potential immunogenicity issues. Recent developments in aglycosylated antibodies possessing similar antigen-binding affinity and engineered-specific FcγR binding has led to an enormous interest in these products. In this article, the authors review the status of expression of antibodies in microbial hosts and present the recent advances in the production of aglycosylated antibodies in bacteria.
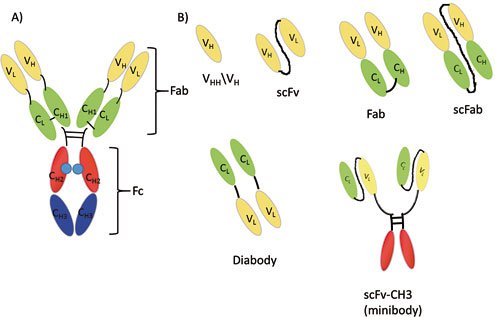
Antibodies are glycoproteins consisting of two heavy chains and two light chains that are linked with disulfide bonds. This Y-shaped molecule possesses an antigen binding fragment (Fab) and a crystallizable fragment (Fc). Presently, different formats of antibody fragments, such as single-chain variable fragments (scFv), the fragment (Fab’)2, diabody fragments (dAb), and single-chain antibody fragments (scFab) have been generated, which possess similar antigen binding affinities, but different effector functions, due to the lack of an Fc region. These different antibody formats have been illustrated in Figure 1.
Motivation for antibody production in microbial hosts
Mammalian cells have been predominantly employed for the expression of antibodies and related products because of their ability to introduce post-translational modifications similar to that of human cells. However, use of mammalian expression system also has few disadvantages, which has propelled manufacturers to search for alternative host systems. One of the most important challenges is predicting the manufacturing behavior of cell lines at early stages and selection of clones with appropriate growth characteristics. Process improvements have generally been expensive and time consuming. Medium complexity, serum requirements, and shear sensitivity are some of the other drawbacks that characterize mammalian systems (5). Cell-culture process parameters affecting the glycosylation level of mAbs also need to be controlled to ensure consistent product quality (6). Selection of a stable antibody-producing clone is also a tedious and cumbersome exercise. In addition, viral contamination of a therapeutic protein preparation can be altogether avoided with cultivation in microbial system. While mammalian systems have been predominantly used for the expression of therapeutic proteins, advances in cellular engineering of microbial hosts have resulted in an increasing acceptance in the use of these hosts as an alternative for the production of mAb therapeutics.
Challenges associated with the production of antibodies in microbial hosts
Microbial cells such as yeast and bacteria possess many advantages, such as fast growth, well-known genetics, and low cultivation costs. Glycoengineering platforms, display technologies and library creation, and robust manufacturing process development support the use of microbial cells as suitable candidates for pharmaceutical development. Although bacterial cells do not have the glycosylation machinery necessary for mAb production, recent successes in the expression of antibody-related products demonstrate the application of a bacterial host as an alternative choice. A number of bacteria, such as Escherichia coli, Corynebacterium glutamicum, Pseudomonas putida, and Bacillus megaterium have been used for the production of recombinant proteins (7). Of these, E. coli is the most popular host and two of the commercially available antibody-fragment products—Lucentis (ranibizumab injection) and Cimzia (cerolizumab pegol)—are expressed in E. coli (8).
Antibody molecules contain disulfide bonds in their structure, and proper formation of these bonds is highly crucial for folding, solubility, and epitope binding. Protein folding and disulfide bond formation require an oxidizing environment, which is provided by the periplasm, the main compartment for the production of functional therapeutic proteins. Co-expression of foldases and molecular chaperones (Skp, FkpA, DsbA, and DsbC) helps in disulfide bond formation, prevention of misfolding, and aggregation of heterologous proteins (9). E. coli has several cytoplasmic endogenous proteases that can cause proteolytic degradation; this represents a critical disadvantage of E. coli as a production vector. However, this problem has been successfully obviated by either use of protease-deficient strains or secretion of protein into periplasm, where there are fewer proteases. In E. coli, the Sec-dependent pathway has been used for protein secretion because of its abundance, although accumulation of protein aggregates may occur due to the generation of premature proteins before secretion. The formation of the inclusion bodies can be avoided by co-expression of cytoplasmic chaperones and utilization of a signal recognition particle (SRP)-dependent pathway. In addition, periplasmic inclusion bodies may also form due to incorrect folding of translocated proteins in the periplasm. Based on the twin-arginine translocation system (TAT), a new transport system enabling translocation of fully folded proteins in bacterial cells has been developed (10). Many advances such as translation region initiation (TIR) optimization, engineering of secretion pathways, and co-expression of molecular chaperones have been made for enhancing antibody productivity in bacteria and these have been reviewed elsewhere (7, 9, 11). Major focus in antibody production in E. coli has been on the generation of scFv and Fab fragments. Full-length antibody (IgG) production continues to be a challenge due to low productivity, and various strategies have been employed to counteract this issue.
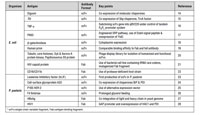
Table I: Examples of antibody fragment production in microbial hosts such as Escherichia coli (E. coli) and Pichia pastoris (P. pastoris).
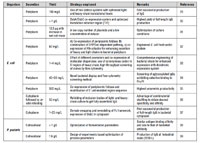
Table II: Production of full-length immunoglobulins in Escherichia coli (E. coli) and Pichia pastoris (P. pastoris).
Aglycosylated antibodies as alternate therapeutic immunoglobulins
Antibodies constitute an important class of serum glycoproteins, and the IgG isotype is the most common isotype used for pharmaceutical applications. Natural IgG antibodies are glycosylated at the Asn297 amino acid position of the Fc fragment. The complex biantennary glycan structure depends on multiple glycosyltransferases and glycosidases located in the endoplasmic reticulum and Golgi bodies. These glycan molecules impart effector functions—such as antibody-dependent cell cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC)—to the antibody molecules. Absence of post-translational modifications in bacterial host cells leads to a complete abolition of glycosylation of antibodies, resulting in structural changes such as open conformation, enhanced flexibility, and complete loss of effector functions. In early experimental studies on X-ray structures of wild-type glycosylated IgG molecules and truncated Fc domains, glycan molecules were considered to provide the necessary flexibility to the CH2 domain of an IgG molecule, and removal of a glycan moiety was thought to be associated with the “closed IgG” conformation (40).
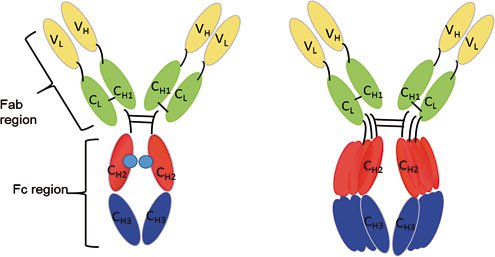

Table III: List of advances in the engineering of effector functions in aglycosylated antibodies. Escherichia coli=E. coli; Pichia pastoris=P. pastoris.
Aglycosylated antibodies are similar to their wild-type glycosylated counterparts in terms of bioavailability, pharmacokinetics, and epitope binding. Several aglycosylated antibodies are under clinical trials and to date, no immunogenicity issues have been reported. Tolerx (MA, USA) has developed a series of humanized aglycosylated antibodies (TRX1, TRX4, TRX518) by knocking out asparagine at the 297th position. TRX1 is a monoclonal IgG1 anti-CD4 mAb (N297A mutation) that is being exploited for the suppression of autoantibodies in autoimmune disorders and neutralizing antibodies induced by enzyme replenishment therapy (44). Onartuzumab (MET-mAb, Genentech) is the first full-length monoclonal humanized and affinity-matured aglycosylated antibody expressed in E. coli for the treatment of lung cancer. It inhibits hepatocyte growth factor (HGF)-mediated activation and receptor signaling and possesses similar pharmacokinetic properties as that of its glycosylated counterparts. Similarly, yeast-expressed aglycosylated monoclonal antibody ALD518 (Alder Biopharmaceuticals) exhibited a half-life of 25 days (comparable to the wild-type glycosylated form) and significant improvements in patients with rheumatoid arthritis (45). Recent success in aglycosylated IgG production and clinical efficacy data indicates that they may have even more applications in the future.
Conclusion
The development of microbial expression systems that are capable of delivering projects that are not immunogenic and retain antigen specificity could pave the way for the cost-effective production of mAb products. Though substantial progress has been made in cellular engineering and in the secretion pathway of microbial cells, further progress in metabolomic and proteomic techniques is required to improve the understanding of microbial systems and the generation of host strains with enhanced potential.
References
1. G. Walsh, Nat. Biotechnol. 32 (10), pp. 992–1000 (2014).
2. J.G. Elvin, R.G. Couston, and C.F. van der Walle, Int. J. Pharm. 440 (1), pp. 83–98 (2013).
3. L. Liu, J. Pharm. Sci. 104, pp. 1866–1884 (2015).
4. G. Kohler and C. Milstein,, Nature 256 (5517), pp. 495–497 (1975).
5. F. Li et al., mAbs 2, pp. 466–477 (2010).
6. A.R. Costa et al., New Biotechnol. 30, pp. 563–572 (2013).
7. A. Frenzel, M. Hust, and T. Schirrmann,, Front. Immunol. 4, pp. 217–236 (2013).
8. C. Zhou et al., mAbs 2 (5), pp. 508–518 (2010).
9. Y.J. Lee, Y. J. and K.J. Jeong, J. Biosci. Bioeng. 120 (5), pp. 483–490 (2015).
10. C.F.R.O. Matos et al., Biotechnol. Bioeng. 109, pp. 2533–2542 (2012).
11. O. Spadiut et al., Trends Biotechnol. 32 (1), pp. 54–60 (2014).
12. H. Li et al., Nat. Biotechnol. 24 (2), pp. 210–215 (2006).
13. T.I. Potgieter et al., Biotechnol. Bioeng. 106 (6), pp. 918–927 (2010).
14. R. Levy et al., Protein Expression. Purif. 23 (2), pp. 338–347 (2001).
15. R. Wang et al., Front. Cell. Infec. Microbiol. 3, pp. 72 (2013).
16. T. Yang et al., Protein Expression Purif. 76 (1), pp. 109–114 (2011).
17. Y. Lee and K. Jeong, Biotechnol. Bioprocess Eng. 18 (4), pp. 751-758 (2013).
18. P. Martineau, P. Jones, and G. Winter, J. Mol. Biol. 280 (1), pp. 117–127 (1998).
19. S. Padiolleau-Lefevre et al., Mol. Immunol. 44 (8), pp. 1888–1896 (2007).
20. P. Philibert et al., BMC Biotechnol. 81 (8), pp. 1 (2007).
21. A. Nadkarni, L.-L.C. Kelley, , and C. Momany, Protein Expression Purif. 52 (1), pp. 219–229 (2007).
22. C. Chen et al., Biotechnol. Bioeng. 85 (5), pp. 463–474 (2004).
23. R. Ridder et al., Nat. Biotechnol. 13 (3), pp. 255–260 (1995).
24. L. Damasceno et al., Appl. Microbiol. Biotechnol. 74 (2), pp. 381–389 (2007).
25. C. Gurkan, S.N. Symeonides, and D.J. Ellar, Biotechnol. Appl. Biochem. 39 (1), pp. 115–122 (2004).
26. N.K. Khatri et al., Biotechnol. J. 6 (4), pp. 452–462 (2011).
27. D. Ning et al., BMB Reports 38 (3), pp. 294–299 (2005).
28. B. Gasser et al., Biotechnol. Bioeng. 94 (2), pp. 353–361 (2006).
29. L.C. Simmons et al., J. Immunol. Methods 263 (1–2), pp. 133–147 (2002).
30. D. Reilly and D. Yansura, “Production of Monoclonal Antibodies in E. coli,” in Current Trends in Monoclonal Antibody Development and Manufacturing, S.J. Shire, W. Gombotz, K. Bechtold-Peters., and J. Andya, Eds. (Springer New York, 2010), pp. 295–308.
31. C.E.Z. Chan et al., PLoS ONE 5 (4), e10261 (2010).
32. Y.J. Lee et al., J. Biotechnol. 165, pp. 102–108 (2013).
33. T. Makino et al., Metab. Eng. 13 (2), 241-251 (2011).
34. S.T. Jung et al., P. Natl. Acad. Sci. 107 (2), pp. 604–609 (2010).
35. Y. Lee, D. Lee, K. and Jeong, Appl. Microbiol. Biotechnol. 98 (3), pp. 1237–1246 (2014).
36. R. Hakim and I. Benhar, mAbs 1 (3), pp. 281–287 (2009).
37. M.-P. Robinson et al., Nat. Commun. 6 (2015).
38. T. I. Potgieter et al., J. Biotechnol. 139 (4), pp. 318–325 (2009).
39. J. Ye et al., Biotechnol. Progress 27 (6), pp. 1744–1750 (2011).
40. S. Krapp et al., J. Mol. Biol. 325 (5), pp. 979–989 (2003).
41. Borrok, M. J., Jung, S. T., Kang, T. H., Monzingo, A. F., and Georgiou, G., ACS Chem. Biol. 7 (9), 1596-1602 (2012).
42. Jung, S. T., Kelton, W., Kang, T. H., Ng, D. T. W., Andersen, J. T., Sandlie, I., Sarkar, C. A., and Georgiou, G., ACS Chem. Biol. 8 (2), 368-375 (2012).
43. W. Shatz et al., mAbs 5 (6), pp. 872–881 (2013).
44. C. Ng et al., Pharm. Res. 23 (1), pp. 95–103 (2006).
45. P. Mease et al., Ann. Rheum. Dis. 71 (7), pp. 1183–1189 (2012).
46. M.-S. Ju et al., Mol. Immunol. 67 (2b), pp. 350–356 (2015).
47. R. Stewart et al., Protein Eng. Des. Sel. 24 (9), pp. 671–678 (2011).
48. S.L. Sazinsky et al., P. Natl. Acad. Sci. 105 (51), pp. 20167–20172 (2008).
49. S.T. Jung, T.H. Kang, and D.-I.Kim, Biotechnol. Bioproc. Eng. 19 (5), pp. 780–789 (2014).
Anurag S. Rathore is a professor, Department of Chemical Engineering, Indian Institute of Technology, New Delhi, India.
Jyoti Batra is a post-doctoral fellow at the Indian Institute of Technology Delhi, New Delhi, India.