Surface plasmon resonance is helping define bispecific antibodies, the next-generation of biopharma therapeutics.
By Robert Karlsson
Abstract
Biotherapeutic antibodies are still the largest growing class of medicines and are used globally to treat a wide range of diseases such as cancer, asthma, and rheumatoid arthritis. Compared to traditional small-molecule drugs, their structures are complex, and they may assert their effects by binding to more than one target molecule. To date, the majority of monoclonal antibodies (mAbs) have generally targeted a particular antigen; however, there are several additional antibody formats currently in the development pipelines of pharmaceutical and biotech companies. One of these promising groups of antibodies is called bispecifics, where one antibody can simultaneously bind to two targets.
Advanced antibody engineering has resulted in the ability to manufacture new recombinant bispecific antibodies suitable for therapeutic use. FDA has approved a bispecific antibody—Amgen’s Blincyto (blinatumomab)—for the treatment of refractory B-cell precursor acute lymphoblastic leukemia (ALL). Blincyto targets cell surface proteins CD19 and CD3 simultaneously, helping to put T cells within reach of the targeted cancer cell with the goal of allowing the T cells to inject toxins into cancer cells, prompting cell death (1). Bispecific antibodies, thus, have the potential to improve cytolytic effects in cancer therapy. Bispecific antibodies are also considered an effective platform for the delivery of therapeutic antibodies across the blood-brain barrier (2). This suggests that in certain cases, bispecific antibodies may have a clinical advantage over monospecific antibodies.
During development and quality control, a range of analytical technologies are used to characterize biotherapeutic drugs, including methods to analyze their structural integrity and activity. Target/ligand binding is an essential critical quality attribute (CQA) that needs to be monitored. The enzyme-linked immunosorbent assay (ELISA) is often used to assess ligand binding; however, it is an indirect method and the output is limited to end-point data. By contrast, label-free technologies such as surface plasmon resonance (SPR) and bio-layer interferometry allow users to follow the dynamics of the interactions so that binding and dissociation events can be directly measured. SPR is a direct binding technique that can measure sequential binding events and is a key part of any analytical toolbox, enabling the examination of dual-target specificities in a bispecific antibody within a single assay.
In the early phases of development (3), the focus is on the drug substance and the assumed mechanism of action. Early biotherapeutic development is typically target based (4), and for a therapeutic antibody, the mechanism of action includes target binding, but may also include Fc receptor and complement binding. A lead substance shall elicit the desired functional response of the target molecule, have adequate bioavailability and biodistribution, and is evaluated from a toxicity perspective.
As the lead candidate enters late-stage development, several CQAs have been established (i.e., properties essential for clinical safety and efficacy), including data on how it interacts with target proteins. See Figure 1 for a sample of the interaction properties that may affect binding activity and concentration of a bispecific antibody.
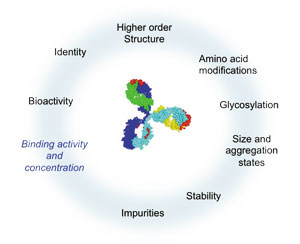
Figure 1: Changes of critical quality attributes, such as high-order structure, amino acid modifications, glycosylation, and stability might impact binding activity and concentration. All figures are courtesy of the author.
Important manufacturing CQAs related to protein integrity, homogeneity, presence of host-cell proteins, host-cell DNA, and/or substances released from process or package material, can be identified using risk assessments. Risk can be assessed by comparing test results with previous experience, knowledge, and through the use of control procedures. While CQA analysis is typically performed in a core analytical lab, there are also other in-process controls that allow a manufacturer to steer the process toward a product with known quality. For each step in the manufacturing process, critical process parameters (CPPs) that can affect CQAs are identified (5). Temperature, pH, cell density, and concentrations of nutrients and metabolites are typical CPPs during cell culture, while other CPPs are important during purification. Controlling these parameters can define the quality of the product. Test procedures and acceptance criteria for CQA analysis have been described in regulatory guidelines for biotechnological/biological products (6, 7).
Late-stage development starts with set up and validation of analytical methods and with securing reagents (8–10) for the analytical program. A broad range (50–60 variants) of different analytical technologies may be used for CQA analysis (11). Mass spectrometry (MS) can be used to establish the identity of the drug (primary sequence) and for detection of size-distribution profiles linked to post-translational modifications such as glycosylation (12). MS is further used in combination with high-performance liquid chromatography (HPLC) and other chromatographic methods to detect and localize amino acid modifications (13). Chromatography techniques are broadly used for detection and isolation of charge and size variants (14). Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) and Western blotting demonstrate protein integrity and in the case of antibodies, the presence of heavy-heavy-light, heavy-light, or light (HHL-, HL- or L-, respectively) chains can easily be detected (15). ELISA and SPR are commonly used for in-vitro confirmation of biological activity by measuring interactions with antigens, receptor, Fc-receptors, or other binding proteins (11, 13). Cell- or animal-based bioassays can also be used to provide information about the complete function of a biotherapeutic drug.
Process development for chemistry, manufacturing, and controls (CMC) (15) includes selection of a cell line for protein expression, development of the culture process, and purification of the drug substance, while formulation includes selection of excipients for the final drug product. During process development, the goals are to obtain high product yield and a good process economy; to ensure that properties related to initial CQAs are maintained; and to secure that the process itself is well controlled and has low impurity levels. Formulations ensure that the final product has proper stability, administration properties, and pharmacokinetic profile. Throughout all steps of late development, initial CQAs are monitored and possible new CQAs are evaluated. Before transfer to manufacturing, the complete list of CQAs will be established, along with methods for their control. Manufacturing and quality control helps ensure the supply of a consistent quality product to the market. CQAs are determined using the “best” knowledge at a given time; they may later have to be revised based on the results of accumulated clinical data.
Manufacturing timelines extending over decades present both challenges and opportunities. A major challenge is to maintain a high and consistent product quality, as manufacturing and analytical technologies evolve, and material and reagents become obsolete. The opportunity lies in improvements that simplify processes and make them more reliable and/or more economic. The demonstration of comparability does not necessarily mean that the quality attributes of the pre-change and post-change products are identical, but based on results and existing product knowledge, it should be possible to predict that any differences in quality attributes have no adverse impact on the safety or efficacy of the drug product (16). Manufacturing changes may be frequent and approval times for changed products can be lengthy (17).
In this article, the use of SPR in the characterization of bispecific antibodies--including understanding mode of action; defining specific target binding and overall antibody activity; and control of their therapeutic development--are discussed. The applicability of SPR in validating potential bispecific molecules for quality and in monitoring multiple binding sites is also discussed.
SPR analysis is focused on ligand bindingLigand-binding assays are key for characterization of biotherapeutic medicines. SPR and ELISA are extensively used in ligand-binding assays. SPR analysis has been used for antibody characterization for more than 20 years. The sample readout from an SPR system such as Biacore (Cytiva) is related to molecular mass and any binding event can be detected without the use of labels (Figure 2). The readout is continuous, which allows for the quality control of the entire binding event and provides opportunities for data analysis based on binding responses obtained at one or several specific time points (report point analysis) or by comparing and even fitting entire binding curves for determination of kinetic and affinity parameters. SPR systems can deliver a direct binding assay focused on relevant interactions and can provide information on binding activities and detect even very weak interactions (18, 19).
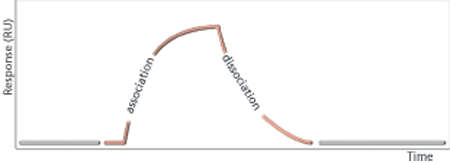
Characterization of binding events is essential for confirmation of CQAs, plays a vital role in forced degradation studies, and can either complement or form the basis for potency assays. Binding assays reflect the molecular mechanism of action and can be developed into such potency assays.
Target binding
Antibodies, cytokines, and hormones typically interact with their receptors. While cytokines and hormones retain their natural sequence and folding, antibody therapeutics are engineered to interact with target molecules (including antigens, Fc receptors, and complement factors) based on their intended mechanism of action. The focus in this section is on the primary target molecule.
Binding properties can vary considerably with estimated dissociation half-lives from 50 seconds (including interferon 2α to IFNAR [20]) to 12 hours (VEGF to bevacizumab [21]). There are currently several antibody formats in development, with a particular focus on bispecific antibodies, where two distinct target functionalities are combined in one molecule. This provides a single biotherapeutic with the ability to target more than one effector function, improving the chance of overcoming or slowing the progression of the disease. More than 20 bispecific antibodies are currently in clinical trials (22). There are two main categories of bispecifics, immunoglobulin G (IgG)-like bispecific antibodies, and small bispecifics. IgG-like bispecifics have a conserved immunoglobulin-constant domain that demonstrate properties of Fc, while smaller bispecifics often lack Fc functionality.
Small bispecifics can potentially reach more hidden targets, but the lack of Fc functionality may result in shorter half-lives and thus, introduce a new balance between half-life and efficacy. In certain cases, pegylation may be used to increase the half-life of these smaller contructs (23). SPR has been used for successful characterization of a number of different bispecifics, including in the following examples:
• Bridge T-cell and target-cell receptors (24)
• Bridge Factor IXa and Factor X (FIXa and FX) binding to mimic the natural function of factor FVIII (25)
• Combine VEGF and Ang-2 functionalities to reduce the formation of blood vessels in cancer tissue (26).
Target binding is clearly an essential critical attribute and has to be controlled during development and later in quality control (QC) for batch-to-batch consistency. While release assays traditionally are based on bioassays, ligand-binding assays (21, 27) can be considered when the mechanism(s) of action is/are defined. Although kinetic data are useful for in-process characterization and batch-to-batch comparisons, release assays require that the ligand-binding assays produce a product concentration that reflects the pharmacological activity (potency assay). Release assays are often based on relative comparisons and parallel line/parallel logistic analyses with defined conditions for equivalence (28).
In one example, the dual specificity assay for Roche’s VEGF-Ang2 CrossMab (19) was developed into a potency assay (26). VEGF was immobilized to the sensor surface, followed by CrossMab and angiotensin injections. In this way, individual binding events of VEGF-CrossMab and CrossMab-angiotensin were measured, as well as the change in binding of angiotension with increasing concentrations of CrossMab. Two response values related to the individual binding events of VEGF-CrossMab (R1) and CrossMab-angiotensin (R2) were obtained. The R2 value that reflects the entire interaction was plotted versus the logarithm of the concentration of the CrossMab. The assay was tested using CrossMabs with known deviation in concentration values including stressed samples, and was shown to have excellent linearity, precision, and accuracy in the range of 60–140% of the nominal concentration.
In another example, a zybody with five specificities was investigated (23). Multivalent molecules, with additional peptides providing functional binding sites to the C- and N-terminii of both the heavy and light chains, were added to adalimumab, trastuzumab, and cetuximab. These zybodies were shown to have increased tumor inhibition and showed improved efficacy in a tumor xenograft model. When different “modular recognition domains” (MRDs) were fused to different chain positions of trastuzumab and analyzed for their binding efficiency using SPR, it was found that fusion of the C-terminus of the light chain resulted in lower binding than at any other potential fusion position analyzed in the study.
Analytical challenges with bispecifics
CrossMab and zybody analyses demonstrate that several specificities in one molecule can be measured in sequence. However, the first binding event may result in a biased selection of antibodies that are available for the second interaction, as antibody molecules with impaired activity may not bind or else bind with lower stability. Therefore, it is important to determine the fraction of active molecules available for interactions. Calibration-free concentration analysis can potentially be used to determine the activity of the separate binding sites. This involves the immobilization or capture of the different antigens, followed by calibration-free concentration analysis (CFCA) analysis. Because bispecific molecules are used as analytes, the ratio of CFCA concentrations can directly provide information about the integrity of the bispecific molecule.
Another assay set up that may prove useful for analysis of bispecifics is illustrated in Figure 3. The bispecific in this example was exposed to two different antigens using a dual-injection protocol. In the first step, the solution from the left tube containing the bispecific antibody saturated with antigen A was injected. Once exposed to surface-bound antigen B, the bispecific antibody bound via the corresponding binding site. Antigen A at high concentration was then injected from the second tube, and dissociation of the antibody from antigen B was monitored in the presence of excess antigen A. This ensures that the observed dissociation is related to the antibody’s interaction with antigen B. This type of experiment can be used to understand if and how the binding of one antigen affects the binding of a second antigen.
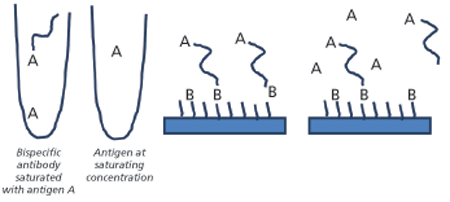
Fc-receptor binding
Fc-γ receptors are expressed on different cell types and can be activating (FcγRI, FcγRIIa and FcγRIIIa), inhibitory (FcγRIIb), or without effect (FcγRIIIb) in antibody-dependent cellular cytotoxicity (ADCC). The mechanism of action for several anti-cancer antibodies involves ADCC, where the interaction with the FcγRIIIa receptor present on natural killer cells may be of particular importance. SPR analysis is widely used in research where antibodies are either designed for improved interaction with Fc-γ receptors (29) or for the elimination of immune effector functions when non-immunostimulatory mAbs are developed (30).
Bispecifics with combined target and Fc receptor functionalities to improve effector functions include HER2/neu antibodies that were combined with F(ab’)s directed to Fcγ receptors (31) or with FcαRI to trigger an F(ab’) directed towards the IgA Fc receptor CD89 (32).
Calibration-free concentration analysis for rapid, absolute, and relative concentration measurements
Concentration analysis based on ligand binding typically requires a standard preparation with known active concentration. A standard preparation may not always be available, however, as is the case when a protein is expressed for the first time or when concentration data for the standard reflects the total protein concentration and not the active concentration. In such circumstances, SPR analysis can be used for the direct assessment of the active concentration. This technique was first described in 1993, but has been further refined with modern numerical integration tools for data analysis (33, 34). The CFCA method illustrated in Figure 4 provides a good estimate of the absolute binding concentration, provided that the diffusion coefficient of the analyte is known and the observed binding rate is flow-rate-dependent. There are small remaining uncertainties related to the conversion of the SPR signal to surface concentration units. All of these uncertainties will cancel out when CFCA is considered as a relative concentration method rather than an absolute concentration method.
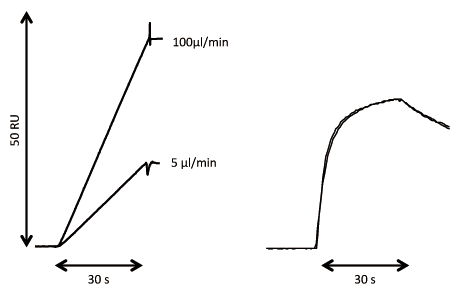
Using CFCA as a relative concentration method is an excellent tool for comparing concentration data, and may be particularly useful for reagent characterization and as a complement to kinetic analysis. This is especially true when changes in the interaction can be related to changes in the active concentration. The use of CFCA for analyzing chromatography fractions and to guide purification efforts have been described (35). Considering that relative concentration data are easy to obtain with CFCA, it may also be used to complement and support potency assays.
Summary
The lifespan of an approved, branded biotherapeutic agent can reach more than 20–30 years. During this time, a manufacturer must develop and deliver a product with consistent quality. To support this process, regulatory authorities such as FDA and the European Medicines Agency have issued guidelines aimed at securing safety and efficacy of biotherapeutic medicines. Antibodies, cytokines, and hormones assert their actions through interactions with their target molecules, and ligand-binding assays are highly relevant for the characterization of these drugs.
Label-free technologies such as SPR can be used in cell culture, purification, and formulation workflows for the determination of antibody concentration and for kinetic analysis of drug-target interactions. SPR measures interactions directly and is capable of measuring sequential binding events. This enables the analysis of dual-target specificities for a bispecific antibody in a single assay set up. SPR has already been applied as a potency assay for bispecific antibodies, and the use of CFCA introduces novel opportunities to assess the activity of separate binding sites for bispecific and multispecific antibodies.
Bispecific and multispecific antibodies constitute a growing area of biotherapeutics. Improving the assessment and understanding of multiple binding sites will be important to understanding their mechanisms of action, to determine their optimal configuration, and for quality control in manufacturing. SPR, therefore, has an important role in the ongoing development of bispecific antibodies.
References
1. Amgen, “Amgen Receives FDA Breakthrough Therapy Designation For Investigational BiTE Antibody Blinatumomab In Acute Lymphoblastic Leukemia,” Press Release, July 1, 2014.
2. Y.J. Yu et al., Sci. Transl. Med. 6 (261), 261ra154 (2014).
3. H.H. Shih, “Discovery Process for Antibody-Based Therapeutics,” in Development of Antibody-Based Therapeutics, M.A. Tabrizi et al. Eds. (Springer Science+Business Media, New York, NY, 2012), pp. 9-32.
4. D.C. Swinney and J. Anthony, Nat. Rev.Drug Discov. 24, pp. 507-519 (2011).
5. M. Mitchell, BioPharm Int. 12 (26), pp. 38-47 (2013).
6. ICH, Q6B Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products, Step 4 version (September 1999).
7. FDA, Guidance for Industry, Bioanalytical Method Validation (Rockville, MD, May 2001).
8. D.M. O’Hara et al., AAPS J. 14 (2), pp. 316-328 (2012).
9. R.F. Staack, et al., Bioanalysis 3 (5), pp. 523-534 (2011).
10. M. Federici et al., Biologicals 41, pp. 131-147 (2013).
11. A. Beck et al., Trends in Analytical Chemistry 48, pp. 1-10 (2013).
12. J. Visser et al., BioDrugs 27 (5), pp. 495-507 (2013).
13. L.C. Santora et al., Anal. Biochem. 299 (2), pp. 119-129 (2001).
14. Cytiva, “Analysis of Therapeutic Antibodies using Amersham WB System,” application note 291140-27 AA (Sept. 2014).
15. S.D. Jones, P. Seymour, and H.L. Levine, Contract Pharma, April 20I0.
16. ICH, Q5E Comparability of Biotechnological/Biological Products, Step 5 version (June 2005).
17. C.K. Schneider, Ann. Rheum. Dis. 72 (3), pp. 315-318 (2013).
18. FDA, Guidance for Industry: Scientific Considerations in Demonstrating Biosimilarity to a Reference Product (Rockville, MD, Apr. 2015).
19. C. Gassner et al., J. Pharm Biomed Anal. 102, pp. 144-149 (2014).
20. C. Dhalluin et al., Bioconjugate Chem. 16 (3), pp. 518-527 (2005).
21. N. Papadopoulos et al., Angiogenesis 15 (2), pp. 171-185 (2012).
22. I. Spasevska, BioSciences Master Reviews. Department of Biology, University of Lyon (January 2014), accessed on Sept. 22, 2015.
23. D.W. LaFleur et al., mAbs 5 (2), pp. 208-18 (2013).
24. P.A. Moore et al., Blood 117 (17), pp. 4542-4551 (2011).
25. T. Kitazawa et al., Nat. Med. 18 (10), pp. 1570-1574 (2012).
26. W. Schaefer et al., Proc. Natl. Acad. Sci. USA 108 (27), pp. 11187-11192 (2011).
27. N. Rieder et al., BioProcess Int. 8 (6), pp. 33-42, (June 2011).
28. USP, USP General Chapter <1032>, “Development and Design of Biological Assays,” USP 35-NF 30:162 (US Pharmacopeial Convention, Rockville, MD, 2010), pp. 1-36.
29. X.R. Jiang et al., Nat. Rev. Drug. Discov. 10 (2), pp. 101-110 (2011).
30. O. Vafa et al., Methods 65 (1), pp. 114-126 (2014).
31. B. Stockmeyer et al., Cancer Res. 57 (4), pp. 696-701 (1997).
32. T. Valerius et al., Blood 90 (11), pp. 4485-4492 (Dec. 1, 1997).
33. R. Karlsson et al., J. Immuno. Methods 166 (1), pp. 75-84 (1993).
34. E. Pol, J. Vis. Exp. 37, pp. 1746 (2010).
35. Cytiva, “Accurate Comparability Assessment of a Biosimilar Interferon in Process Development,” application note 29-1154-78 AC (2014).
About the Author
Robert Karlsson is senior staff scientist of protein analysis applications at Cytiva.